第二届网络作品 使用液质联用仪建立抗生素多残留检测方法的体会 电子版
第二届网络作品 使用液质联用仪建立抗生素多残留检测方法的体会 电子版zhufangwei
第2楼2009/08/29
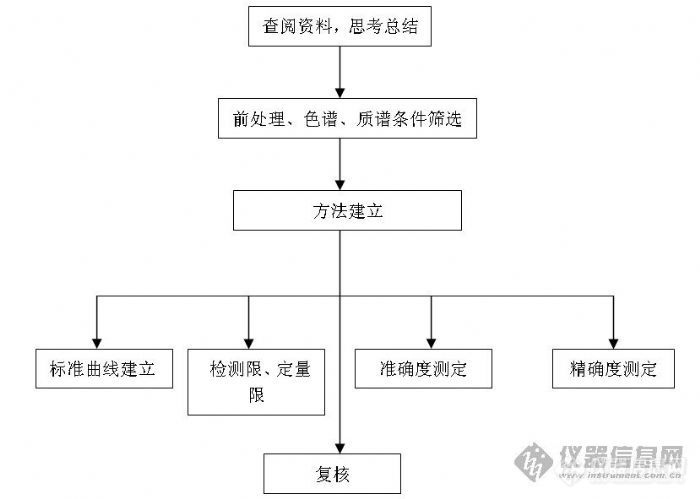
如何去完成这样的工作呢,要找到创新点才会更有做这样研究的必要和意义,就必须在广泛查阅文献的基础上,主要是那些英文的相关文献了,这起码就是一个长期的工作,不仅要看,而且要总结归纳,包括在整个试验的前期、过程和后期,不断的查阅资料都是必须的。多残留检测就是一个很大的创新点,一个药物一个药物的检测方法怎么都不如同时检测一类这样的药物来得快,并且节省成本,省事。一般同一类抗生素是会包括很多药物的,如何选择检测药物也是牵扯到很多因素的,查阅国家规定的法规是应该的,需要确定的是药物的残留标示物,有一些药物检测的并不是原形,并且有的药物残留标示物还不止一个。在我国很多规定还是与欧盟法规接近的,包括方法建立过程需要涉及到得指标,很多人都知道什么回收率啊,变异系数,线性范围,定量限等等,在不同浓度范围规定的数值都是不一样的,所以要做到心中有数,建立方法首先要有个依据和知道要达到什么样的结果。
zhufangwei
第4楼2009/08/29
质谱那块优化的一些参数就是比较固定的,这些对于一个初学者来说就是可以通过前面使用过仪器的人获得的,以后再慢慢的去理解为什么要优化这些参数,意义在哪里,这都是需要查看自己所使用的仪器很多由点到面的资料才会达到的档次。在进行质谱优化时,一定要保证基本的正确操作,因为这是第一步,如果不能保证的话下面的也没有必要做下去了,因为做出来的东西也完全是误导。做残留检测,要求一般的是一个母离子对应两个到三个子离子就可以达到要求的鉴定点数了。首先要保证母离子的强度,然后在进行三个最强最稳定子离子的确定。有时候会发现找不到分子离子峰的情况,可能有一些加和离子出现。这样的情况可能就需要改变流动相的成分了,这个时候也只是为了质谱优化强度做的努力,与后面色谱条件的确定还不相关。一般都是可以选择到M+1峰的,是在选择不到也只能用加和离子了。
zhufangwei
第5楼2009/08/29
在确定好质谱那块的条件之后就可以作为一个检测条件编入仪器的文件方法里面了,相当于一个检测器的条件吧,然后就是色谱条件的优化,一般会选择很多流动相的添加剂来做比较,做多残留检测方法最好用梯度洗脱了,一般设置一个样的时候在20-30分钟,色谱条件主要还是看有无保留,峰型怎么样,如果没有保留的话首先想的是更换流动相,一般对于反相色谱C18基本可以满足要求,除非是那些特别特殊极性很强、有特别要求的药物,比如三聚氰胺等要使用特殊型号的色谱柱,通过流动相添加剂的选择是可以大大的提高检测物的响应的,尝试不同的梯度来保证峰型和将同一类的药物分开,达到最理想的状态。虽说液质可以不要求那么严格的分离,但是之间如果分开不好的话也会药物和药物之间有离子抑制的,主要表现在离子源那块电离的时候,如果在色谱柱上不保留的话也很容易受到杂质的干扰,因为一般很多杂质不保留都是在那个时候出来的,对检测物是会有很大响应的,包括什么基质效应,杂质干扰就可以在这种情况下显现出来。其实试验稍微做的有创新一点,能够保留是个很重要的坎,比如同时检测非极性的药物和极性的药物,很难找到一个理想的条件,这就必须要多多尝试了,因为很多药物的残留标示物是其代谢物,是极性很强的,没法避免的。
zhufangwei
第6楼2009/08/29
如果质谱优化和色谱优化你都能保证了,下来才有幸做组织样品的前处理方法了,这个应该算是最重要的一个步骤了。查阅文献相当重要的目的也是为了为建立方法确定一个好的前处理方法做一个参考的依据。前处理很起始的就是要对回收率进行考察,当然要求前处理的步骤要越简单越好了,设计要合理。一般采用液液萃取和固相净化相结合的方式,选择有机试剂提取也是根据检测目标物具体的理化性质来决定的,这些都是在文献调研时应该充分做的工作,有些是会涉及到使用混合提取液来进行的,固相萃取柱的选择也是很有讲究的,目前有很多种不同型号的固相萃取柱,常用的是非极性吸附的C18,也有阴阳离子交换柱,混合阴阳离子交换柱等等,这也都是与检测药物的理化性质相关的。