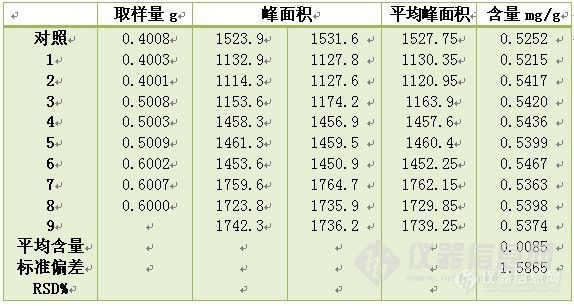
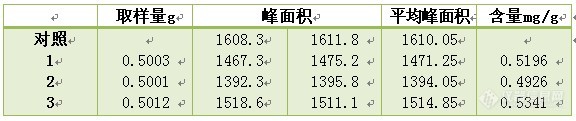
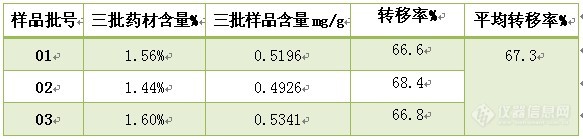
titi
第1楼2009/12/10
此楼为版友提问汇总区:(不断更新中)
1.一种新药的含量侧在HPLC检测中,供试品浓度的选取应该遵循什么的原则?浓度多少合适?以定量限做浓度检测的依据吗?10倍的信噪比?遵循最大还是最小浓度原则?
2.做回收率的时候,是取了9分不同含量的样品。我看到有的回收率是取3分不同含量的样品,分别加入其80%,100%,120%.也有的取6分不同含量的样品,加入其80%,100%,120%的对照品。请问:回收率有统一的取样标准吗?
3.关于含量低于万分之一时需要配合别的指标这条原则的理论依据是什么?或者说出发点是什么?怎么说它至少也是一个半定量指标,总比纯粹的鉴别指标好吧?
4.“如含有苷类成分的新药,如采用水解后苷元的含量为含测指标则难以反映在贮存期问苷类成分水解成苷元的情况。”如果是以皂苷为指标成分,为了防止找储藏过程中皂苷水解的问题,应该怎么做?对于成品只做稳定性试验,能说明问题吗?
5.许多中药有效成分测定含量要用外标法测定,这有一个问题。许多有效成分很难有法定机构来源的对照品,买一些试剂公司的试剂可以吗?对方有本公司的报告。
6.请问提取溶剂考察的时候只用作一份样品就够了吗?就我所知,好像要做2份才对啊,因为你不好排除是不是偶然误差啊!
7.2010版药典增加了不少HPLC检测,以前用UV或滴定检测方面的产品现在都改液相了,请问,这种检测方法的转变是应该以产品特性区分对待,还是以HPLC检测作为常量分析的主流?
8.请问在做专属性时 ,一定要做酸碱氧化破坏吗.
9.如果一个西药(是从一位中药里面提取的单体,由于同分异构体太多,现在想改为西药注射液),现在想要重新申报成中药的话,标准需要做哪些工作?
10.中药成分比较复杂,提取标准品或对照品给新药审评的时候,目前要求纯度达到99%吗?现在都使用液相色谱法或毛细管柱分析,特别是挥发性成分,使用毛细管柱色谱法,能达到95%的含量已经是很不容易的了。
11.打一个不太实际的比方:如果皂苷是指标成分,但是降解后的苷元可能有一定的副作用。在做长期试验中,0个月为2.0%,两年后为1.6%;标准为1.5%合格。在这种情况下,就指标成分来说是合格的,但是降解后的产物算是杂质吗?
如果降解后的产物有一定的副作用,在新药申报时,国家对这个有没有一定的要求?
12.还有几个问题要请教:
(1). 在申报复方中药时,在方法验证过程中,对药材是不是也要进行验证?药材的检测方法都是按照药典要求的。如果要验证,都要做哪些?
(2).在耐用性检查时,流动相比例变化、检测波长、色谱柱厂家等,有没有硬性规定一定要变,最少变动几个因素就可以了?
如果波动,流动相变动和检测波长变动的比例在多大的范围能说明问题;色谱柱最少要换几个?
(3).我用ELSD检测,像蒸发温度、气体流速是不是也要变动?范围是多少?
(4).在变动每个因素之后,是不是都要进行重复性验证?
13.我们已经通过GMP验收了,明年想加一个新药进行投产,临床试验也通过了。正好明年又要进行GMP的一次验收,请问我们对新药的验收要做哪些具体的工作呢?稳定试验等都需要有申报材料吗?具体希望给我们一个思路来。
14.你好!我有两个问题。感谢,期待你回复!1.看了你的这个回答,我感觉每份进两针的意义不大,因为色谱仪器的稳定性保证了你一份样品进2针的结果是一样的。2.我想问一个关于含量测定中有关对照品样品数量的问题:
在进行含量测定时,我们应该选择几份对照品呢?
a.如果1份,应该进几针?结果RSD要求为多少?
b.如果2份,应该怎么进针?结果RSD要求为多少?
c.其它情况的话是怎么样的?
15.首先感谢xy4585618的答复,先还有个问题:就是在开始含量测定前,需要对仪器校正一下吗?如进样看看重复性、看看线性等?
16.你好!我们正在做一个十几味复方中药的质量标准。
(1).日间精密度不需要做吗?
(2).现在中药含量限度的制定都要求做十批样品,至少二十个数据来确定。因为中药的批间差异太大了。
(3).标准品的纯度必须做吗?我的标准品里有杂质,但是没有做纯度检查,含量就按买来的时候表示的97%的量计算。
(4).中药的回收率和化药有什么差别吗?化药做加样回收率时,样品也是减半吗?
17.感谢版主的回答,我看了一下“《中国药典》中药质量标准研究制定技术要求”规定含量限度,要求测定十批,并没有临床和生产之分啊。不知道在哪可以查得到,对于报批临床和生产要求不一样吗?我以前倒是没有注意过这一点。我的标准品是在中检所买的,包装上写着:供含量测定用,以97%计。中检所卖给我们的标准品,本身就达不到98%的纯度。另外,本想问,不需要做中间精密度吗?即不同日期、不同分析人员、不同设备、仪器对结果的影响
18.疑问:“加样回收试验即于已知被测成分含量的成药中再精密加入一定量的被测成分纯品,依法测定。用实测值与原样品中含测成分之差,除以加入纯品量计算回收率。此法不用制备空白对照,模拟真实性好”
请问:“已知被测成分含量的成药”你是如何已知的?你是否先用你所要验证的方法先测样品的含量?若是这样,这中间会不会存在逻辑套用的错误?
19.刚接触,新药含量测定研究的大概流程是怎么样的,一般都是参考那类的资料啊
20.请问一种新药的含量测定是否可以采用新的分析方法如芯片电泳等技术?如果采用新的技术手段获得的结果在某一家生产厂家获得了结果是否就可以申请相关的国家标准?
21.2010板药典中,很多都需采用液相测定含量了,请问版主,滴定法测定含量与液相那个比较精确
22.若新药含量未知,在HPLC检测中,标准品浓度的选取应该遵循什么的原则?一般选择什么浓度范围何时呢?若标准品液相时出现杂峰,那么定量时要不要考率杂峰的影响?谢谢!
23.做新药申报的话,含测还应做中间精密度试验(变换操作人员、试剂、仪器等条件不同日期测定同一样品),耐用性试验(考察流动相比例、柱温变化、pH等微小变化对分析方法的影响)
24.请问版主 如果精密性试验很好 稳定性是否可以省略呢
25.现在对照品都是要求配两个,每个对照的峰面积除以对应的浓度f=A/C,f1/f2 应该在0.98-1.02之间,保证配置的准确性。要是配置的对照品超过了0.98-1.02的范围,应该重新配置还是两个都用?如果在范围之内,用一个对照品能说明问题不?
26.这个问题我也想问一下,
(1).就是在配置2个浓度时,到底相差多少为好?
(2).如果规定是30mg,那么实际称量中这个范围定在多少为好呢?这个范围是否写在文件里呢?
(3).如果在方法中第一次设定称样量,这个称样量设定多少合适呢?30mg、60mg?还是怎么样呢?
27.请问:转移率是什么意思,能详细解释一下吗?
28.xy4585618您好!我有以下问题想请教:
1.请问西药的有关物质检查中破坏性实验的条件一般是怎么确定的,中药的成分已经很复杂了,还用不用做破坏性实验?
2.有关物质检查中,杂质是不是只用定量就行了,什么情况下需要确定杂质结构呢,杂质的定量一般用什么方法?
29.还想请教一下,在HPLC测定某成分时,要先用对照品进行紫外扫描,以确定最大吸收波长,如果该波长与药典上规定的有出入的话,是以紫外扫描的为准,还是药典规定的为准呢?
30.如果主成分与多个已知杂质在最大波长紫外吸收灵敏度相差较大,在适当改变波长(对主成分含量测定影响不大情况下),能大大提高杂质的检测限和定量限,是否可以不用最大波长 ?
31.在研究注射药品的稳定性时,考虑到从配置到注射会有一段时间,尤其是在静脉滴注时。那么,我们在方法考察时,是否要对注射剂溶解后样品进行检测稳定性呢?
32.就是在做方法验证的时候,信噪比的范围该如何啊?有没有一个比较法定的依据?具体该选多大的范围,在什么地方选取?
33.做残留抗溶剂(如产品M中的乙醇含量)方法验证时,是否做所配制溶液的稳定性,若要做,是做产品M的稳定性或是乙醇的稳定性?谢谢!
34.请问:水溶性2-氨基苯酚-4-磺酸氨基值及盐份检测方法
titi
第2楼2009/12/10
titi
第3楼2009/12/10

titi
第4楼2009/12/10