液相色谱(LC)
dickwang2008
第1楼2009/12/31
问下楼主标品、原药是用什么溶剂来配制的?
wonboy
第2楼2009/12/31
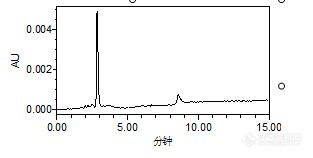
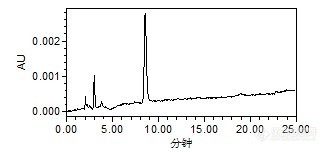
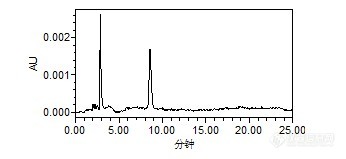
原药和乳油都是先用甲醇配制成100ug/ml的储备液,之后再用流动相稀释至0.2。
有水有渝
第3楼2009/12/31
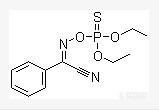
辛硫磷溶解性:不溶于水,溶于丙酮、芳烃等化合物外观与性状:纯品为浅黄色油状液体。1.不能与碱性物质混合使用。2.辛硫磷见光易分解。能不能再去弄另外一家的原药看看是不是一样的情况。
歌名
第4楼2021/06/07
楼主方便把标品“中国标准技术开发公司”网址或者链接贴上来吗?还有原料药的链接(曾经买到过假的原料药),这种固体标品最好去国家标准物质网购买。辛硫磷GC测的方法做的多,LC没做过,看你说的分析方法应该是没问题,辛硫磷最大吸收波长在254nm左右,你看一下查看一下你的图谱2.9和8.5min最大吸收波长分别是多少?
品牌合作伙伴
执行举报