


wmj31 2012/06/26
[quote]原文由 [b]hansjuan(hansjuan)[/b] 发表: wmj31老师您好!我有以下问题想请教 请问:做无机杂质分析方法验证的时候,还需要做专属性、耐用性和系统适用性这三项吗?方法的LOD应该怎么做?[/quote] [img]http://ng1.17img.cn/bbsfiles/images/2012/06/201206262315_374575_2203990_3.jpg[/img] 我们的做法,对于无机杂质的限度测定,就做下专属性,其实就是加标回收。对于无机杂质的定量测定,火焰就单做耐用性,石墨炉对于专属性、耐用性和系统适用性这三项都没做。方法的LOD拿样品空白进20针,然后3倍吸光度的标准偏差除于斜率就可以得到检出限。
秋月芙蓉
第1楼2012/06/24
药品中无机杂质限度的制定及分析方法验证(一)
杂质是指药物在生产或贮藏过程中引入的,无治疗作用或影响药物的稳定性和疗效,甚至对人健康有害的物质。根据ICH Q3a的定义,杂质可分为有机杂质,无机杂质和残留溶剂。而制药工业中无机杂质的可能来源于生产过程,它们一般是已知的和确定的。其常见来源:1.催化剂;2.原材料;3.辅料(填充剂,稳定剂等);4.生产设备,反应釜,管道等。无机杂质超标引起的危害就不细说了。
本文将分为两部分,第一部分是说中国,欧洲及美国药典对于药物中无机杂质的限值要求,第二部分是讲药品中无机杂质的分析方法的验证。
第一部分 各国药典中无机杂质的限值
鉴于无机杂质超标后可能引起的危害,各国药典都对药物中无机杂质制定了相应的限度,下面将介绍中国药典,欧洲药典及美国药典对于无机杂质这块的一些情况。
一、中国药典
目前2010版中国药典并没有针对各个元素制定相应的限度,只是对重金属检查法制定了限值,一般来说用于口服的原料药控制在小于百万分之20,用于注射用的原料控制在小于百万分之10等。重金属指在pH3.5的醋酸盐缓冲液的条件下能与显色剂(硫代乙酰胺试夜)或硫化钠作用的所有金属盐类的总称。包括银、铅、汞、铜、镉、铋、锑、锡、砷、镍、钴、锌盐等,因铅的显色反应较明显,并且生产中遇铅的机会较多,又易在体内积蓄中毒,因此检查时多以铅为代表。
重金属测定法的一些不足:①不具备元素特异性,所有显色的硫化物都考虑在内。②使用干法处理样品时,特别是经过炽灼残渣检查法后又用于重金属检查的,大量挥发性元素损失。③重现性差,反应物不稳定,随时间变化。④视觉对比的误差。
二、欧洲方面
欧洲药典目前还没有实施针对各个元素制定相应的限度的指导文件,目前也用重金属检查法,但欧洲药物管理局(European Medicines Agency)(EMA或EMEA) 下属的人用医疗产品委员会CHMP(Committee for Medicinal Products for Human Use)制定了关于金属催化剂或金属试剂残留量限度规定的指导文件,其生效日期是2008年9月1日,将在5年后实施。该指导文件的目的是为原辅料和制剂中残留的金属催化剂或试剂提供最大可接受浓度限度。
该指导文件基于安全考虑(对人体健康的潜在风险),将14个金属元素分为以下3类:
第一类金属:具有显著安全性担忧。这一类金属具有已知或怀疑的人体致癌性,或者具有其他显著的毒性。目前第一类的金属还可以再细分为1A、1B和1C三个亚类。
第二类金属:具有低安全性担忧。这一类包含了对人类毒性较低的金属。通常可以耐受此类金属在常见药物中的暴露量。它们可能是营养需求的微量金属,常存于食物原料或营养补充剂中。
第三类金属:安全性担忧最小。这一类包含那些无严重毒性的金属。它们的安全概况已经得到很好的确证。在远大于常见药物中的量时,也可以较好地耐受。通常广泛存在于环境、植物和动物中。
表1是目前通过口服、注射和/或吸入所接触到的14种金属残留的PDE和浓度限度。1A和1C类的接触限度和单个金属有关,1B类的限度和所列出的金属总量有关。对于1B类的铂族金属,之所以采用保守的方法是因为目前的毒性数据很有限。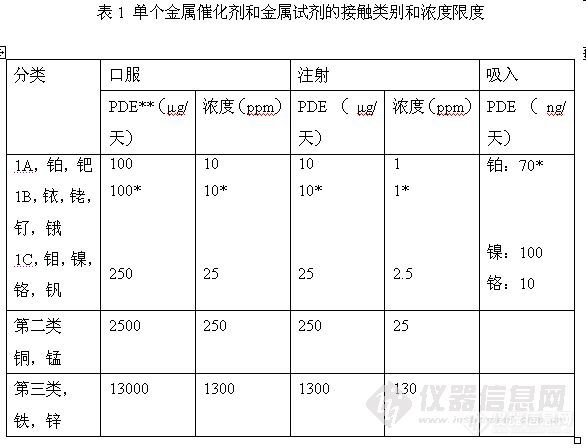
*亚类限度:所列出的金属总量不得超出指定限度
**PDE:日允许接触量
那我们如何根据表1中的来制定原辅料及制剂中金属残留浓度限度,对于这个问题,我们首先要解决的是哪些元素需要测,其残留浓度限度又是多少?
一般来说如果确定或怀疑因为在原辅料的合成工艺中使用特定金属催化剂或者金属试剂而导致金属残留的话,那么必须对每种特定的金属残留制定浓度限度和确证检测,也就是说不是每一个元素都做,只有用到才会去测。比如说,同一种药,A厂中用到镍做催化剂,B厂用到钯做催化剂,那么A厂只要做镍的检测,对镍的残留做要求,而不必对钯做要求,B厂则反之。解决了哪些元素需要测,那么其残留浓度限度又如何制定呢。
设定金属残留浓度限度有两个方法可供选择。
方法1:假设制剂的日剂量小于10g时,每种金属,可以使用表1中的ppm浓度限度。也可以用以下公式,得出其浓度限度。
浓度(ppm)=PDE(μg/天)/日剂量(g/天)
如果制剂日剂量超过10g/天,则使用方法2。
方法2:如果制剂日剂量超过10g/天,则用以下公式,得出其浓度限度
浓度(ppm)=PDE(μg/天)/日剂量(g/天)
可以使用表1 中的PDE(μg/天),结合制剂中原辅料的实际每日给药量,计算出原辅料的金属浓度限度,然后根据药物制剂的每种成分中残留的金属含量叠加起来,每天的总量应低于PDE给定的值。
三、美国药典
美国药典重金属测定法现在还是用USP231进行测定,USP在2009年4月的会议上,承认USP231的不足,并寻求替代方法,将由USP232(元素杂质限值)和USP233(元素杂质测定方法)代替,现就对我了解到USP232中元素杂质限值的一些情况说个说明: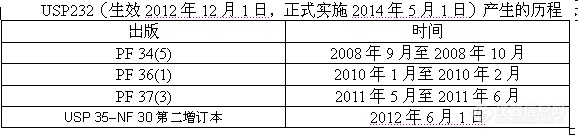
注:药典论坛(Pharmacopeial Forum,PF) 是一份双月刊在线杂志,USP 将其作为开发和修订美国药典和国家处方集 (USP–NF) 标准的平台,通过该平台征求公众的意见和建议。
1. PF 34(5)
药典论坛PF 34(5)首次提出各元素杂质的限度要求,如下表:该元素限度要求只是用于原料药,制剂及辅料,膳食营养剂另做要求。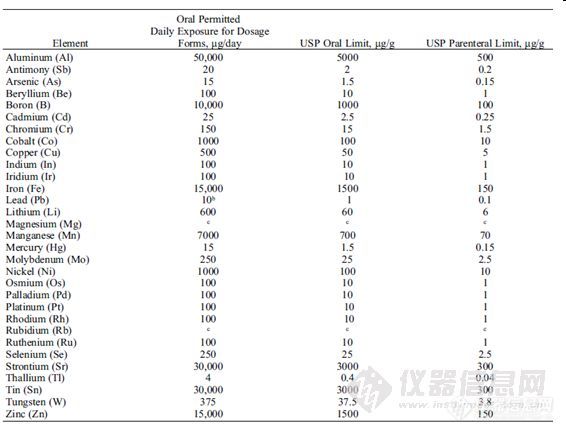
这与EMEA下属的人用医疗产品委员会(CHMP)提出的关于金属催化剂或金属试剂残留量限度规定的指导文件相比,元素更多(31个元素),EMEA指导文件(14个元素),而且主要是增加了As,Hg,Cd,Pb有毒元素的限度。
2. PF 36(1)
PF 36(1)对PF34(5)的草案进行了修改,以下几点是不同的地方:
①把16种元素分为两类
(1)第一类元素杂质:应避免的元素杂质
为人类已知或强烈怀疑的毒物或者环境危害物,其分别为Pb,Cd,Hg,As。
(2)第二类元素杂质:应限制的元素杂质
毒性低于第一类元素。分别为Cr,Cu,Mn,Mo,Ni,Pd,Pt,V,Rh,Os,Ir,Ru。
②PF36(1)中的元素有16个,与PF34(5)相比,少了15个。具体的元素限值如下: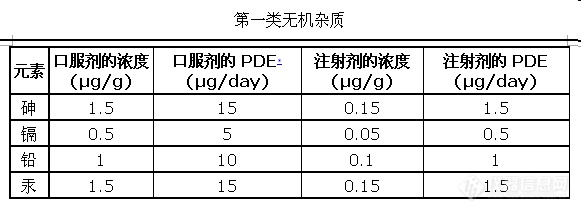
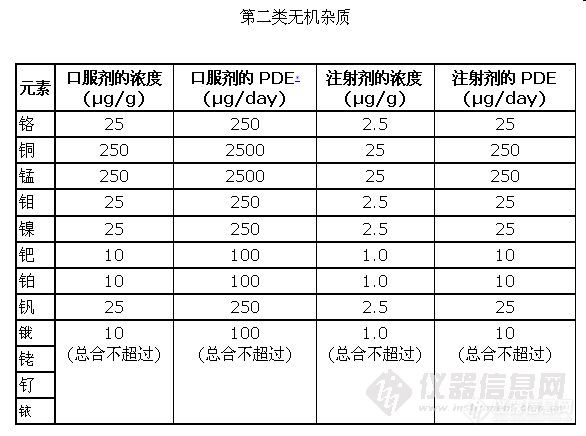
秋月芙蓉
第2楼2012/06/24
3.PF37(3)
PF37(3)对PF 36(1)进行了修改。以下是几点不同的地方: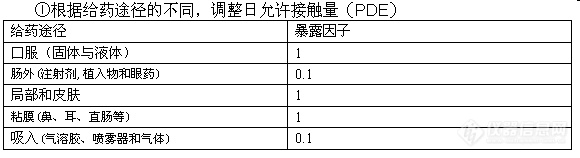
调整日允许接触量(PDE)=日允许接触量(PDE)×暴露因子
②一类和二类金属元素不再列出。制剂和原辅料单独列出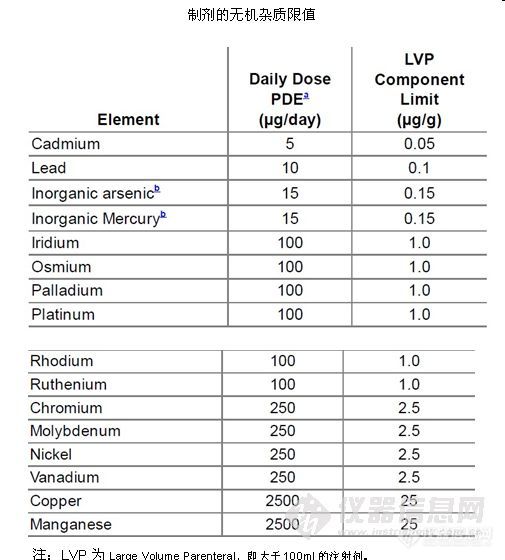
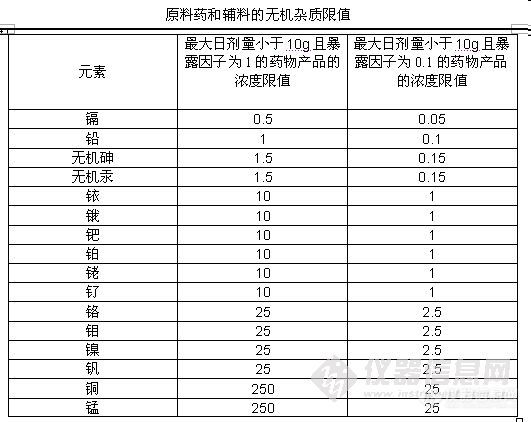
③砷和汞分别修改为无机砷和无机汞,而且无机砷和无机汞超过限值时,要做形态确认。
4 .USP232最终稿
USP232最终稿又分别PF37(3)进行了修改,重点内容如下。
①为了和EMEA指导文件保持一致,取消了暴露因子的概念,根据给药途径分为3类单独列出,分别为口服(oral),肠外(parenteral),吸入(inhalational)。粘膜(mucosal)和局部(topical)的限值等同于口服。
② 无机砷和无机汞超过限值时,要做形态确认。四大毒素必测:铅,砷,汞,镉。
③制剂和原辅料单独列出元素杂质限值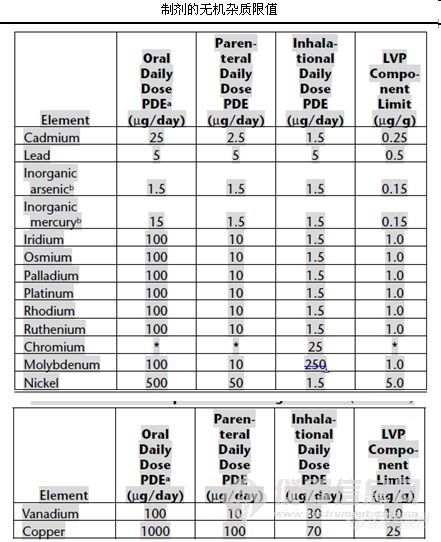
注:(1)LVP为Large Volume Parenteral,即大于100ml的注射剂
(2)钼那一栏中有个错误,吸入剂的限值应为10微克/天,而不是250微克/天。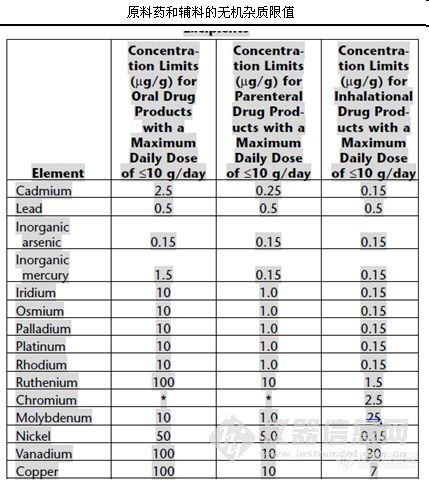
注:钼那一栏中有个错误,吸入剂的限值应为1.0微克/克,而不是25微克/克。
④因为毒胶囊的,而引起关注的铬,因为没有更多的安全性数据,而取消了口服和肠外的限度要求,只是增加了吸入剂中铬的限度要求。锰的限值也取消了。
wmj31
第10楼2012/06/25
让影子说成是老师,感觉怪怪的,呵呵。
文中有写到。USP232生效是21012年12月1日,预计实施是2014年5月1日。你可以到这个看看,http://www.usp.org/usp-nf/hot-topics/elemental-impurities

































