原子吸收光谱分析的定量方法
原子吸收光谱分析室一种动态的分析方法,用校准曲线进行定量。常用的定量方法有标准曲线法、标准加入法和浓度直读法等。如为多通道原子吸收,可以用内标法定量。这些方法中,标准曲线法最为常用。
1、 标准曲线法
原子吸收光谱法是一种相对的测定方法,不能由分析信号的大小直接获得被测元素的
含量。需要通过一个关系式将分析信号与被测元素的含量关系联系起来。校正曲线就是用来将分析信号(即吸光度)转化为被测元素含量(或浓度)的“桥梁”。
校准曲线的质量直接影响样品测定的准确度。因此,校正曲线设计合理。从校准曲线置信范围考虑,当实验点数目少于4点时,置信系数较大且变化速率快,置信范围较宽,由校准曲线求得的含量值或浓度值的不确定度较大。随着实验数目的增加,置信系数减小的速率很慢,置信范围变小的速率很慢,因此用4-6个实验点绘制校正曲线是很恰当的。在总测定次数相同的情况下,多设置实验点,减少每个实验点的测定次数,比少设置实验点,多增加每个实验点测定次数更有利。因为增加某个实验点的测定次数只能改变此点的精密度,而增加实验点数目却可以改变校准曲线的精密度。从测定误差考虑,校准曲线中央部分的精密度优于其两端,因此应让被测元素含量位于校准曲线的中间。
用“空白”溶液校正仪器零点,实际上就是用一个测定误差较大的点做为基准点校正仪器,这显然是不合适的;用空白溶液的测定值直接校正空白值也是不可取的,因测定值是随机变量,而且测定误差较大;用一次测定值作为基准对空白进行校正带来很大的偶然性,校正效果得不到有效的保证。正确的方法是用校准曲线的截距来校正空白,或者对“空白”溶液进行多次测定,取其平均值来校正空白(如下图所示,每次测定的空白是有区别的,且测定结果RSD很大)。

原子吸收光谱法分析是一种动态测量,测定值易受实验条件的影响,引起校正曲线转动或平移,或者既产生转动又产生平移。因此,应该随时或者定时检查校正曲线是否发生了变化。如何检查这种变动?不少分析人员采取的做法是重新测定个别实验点的吸光度,根据新测得的吸光度,将原来的校正曲线平移;或者通过新吸光度值和坐标原点重新制作标准曲线,重置标准曲线斜率。这两种做法都是有问题的。前一种做法将曲线平移,实际上是假定各实验点的偏移大小是固定的,不随被测元素含量而改变,是一个固定系统误差。后一种做法是将斜率重置,实际上是认为曲线的原点是不变的,不存在固定系统误差,只是随被测元素含量而改变的相对系统误差存在。而事实上,固定系统误差和相对系统误差经常是同时存在的,使校正曲线既产生转动又产生平移。校正曲线的正确校正做法是用原来制作校正曲线时不同含量或者浓度的标准样品,测定吸光度,将原有点的与新的实验点合并起来重新绘制校准曲线,既利用了原校准曲线的信息,又利用了新获得的信息。其所以用不同的含量或浓度的标样来检查校正曲线,是因为用原有的实验点与新实验点合并绘制校准曲线时,增加了实验点的数目,有利于提高新校正曲线的稳定性(原始结果如上图所示)。

2、 标准加入法
标准系列与样品的基体精确匹配是制备良好校正曲线的必要条件,分析结果的准确度直接依赖于标准样品和未知样品物理化学性质的相似性。在实际分析过程中,样品的基体、组成和浓度千变万化,要找到完全与样品组成匹配的标准物质是有困难的,特别是对于复杂基体的样品就更加困难。试样物理化学性质的变化,引起喷雾效率、气溶胶粒子粒径分布、原子化效率、基体效应、背景和干扰情况的改变,导致误差增加。标准加入法可以自动进行基体匹配,补偿样品基体的物理、化学干扰,提高测定的准确度。
操作如下,分取几份等量的被测试样,在其中分别加入0,c1,c2,c3,c4,c5等不同量的被测元素标准溶液,依次在标准条件下测定它们的吸光度值Ai=(i=1,2,3,4,5),制作吸光度对加标量的校正曲线,校正曲线不通过原点。加入量的大小,要求c1接近于试样中被测元素含量c0的两倍,c2是c0的三到四倍,c5必须仍在校正曲线的线性范围内。从理论上讲,在不存在背景吸收或校正了背景吸收的情况下,如果试样中不含被测元素,校正曲线理论应通过原点。若校正曲线不通过原点,说明试样中含有被测元素,其含量的多少与截距大小的吸光度值相对应,将校正曲线外延与横坐标相交,原点至交点的距离,即为试样中被测元素的含量c x。
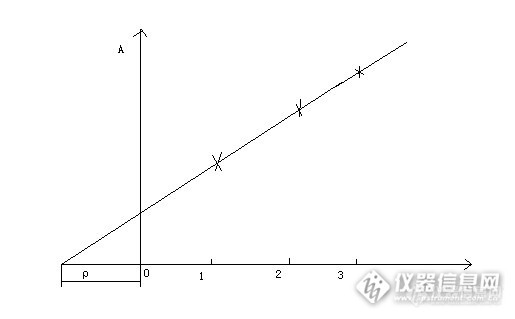
标准加入法所依据的原理是吸光度的加和性。从这一原理要求,不能存在相对系统误差,即试样的基体效应不得随被测元素含量对干扰组分含量比值改变而改变;必须扣除背景和空白值;校正曲线是线性的。
3、浓度直读法
浓度直读法的基础是标准曲线法。先用一个标样定标,通过标尺扩展,将测定吸光度值调整为浓度值,以后测定试样时直接得到它的浓度值。该法的实质就是由该定标点与原点制备校正曲线,预先存于仪器内。以后只要测定试样的吸光度,仪器将自动根据已存的校正曲线算出试样中被测元素的浓度,并显示在仪器上。浓度直读法测定的准确度,直接依赖于校正曲线的稳定性,且要求测得的试样吸光度值必须落在校正曲线上。浓度直读法定量的准确度要逊色于标准曲线法和标准加入法。其优点是快速。
4、试样稀释标样法
先测定体积为Vs、浓度为Cs的标准溶液的吸光度As。在用一定体积Vx的样品溶液稀释标准溶液,稀释后试液的浓度为C=(CsVs+CxVx)/(Vs+Vx),其中Cx是被测样品溶液的浓度。测得稀释后试液的吸光度为Ax,则有以下关系式:
As=KCs
Ac=K(CSVS+CxVx)/(Vs+V)
Cx=Cs[Ac/As(1+Vs/Vx)-Vs/Vx]
在标准条件下,选定了已知浓度Cs的标准溶液,测定了稀释溶液的吸光度值As是一定的,只要用样品标准溶液按照一定的比例稀释标准溶液,测得了稀释标准溶液的吸光度Ax,就可以计算出样品溶液的浓度Cx。
正确选择标准溶液的浓度和稀释比是关键。对于足够低浓度水平的元素,所选择的标准溶液的浓度要是适当高于样品溶液的浓度,稀释比适当,以便能准确的测定稀释后溶液的吸光度值。试样稀释标样法的优点是,用测定较高浓度溶液的吸光度代替测定低浓度水平溶液的吸光度,有利于提高测定吸光度的准确度和精密度。
5、内标法
内标法最大的优点就是可以减少实验条件变动所引起的随机误差,提高测定的精密度。为此,要求内标元素与被测元素具有相同的或相似的物理性质和化学性质;内标物质和被测组分的响应信号易于分辨且不影响被测组分的响应信号;内标物质中不得含有被测元素。因为要同时测定被测元素与内标元素的吸光度值,因此必须要用多通道原子吸收光谱仪器。
内标法是在标准试样和被测试样中分别加入一定量的内标元素,在标准条件下测定分析元素和内标元素的吸光度比Ai/An,以Ai/An对Ci(i=1,2,3,4,5)绘制校准曲线。在同样条件下,测定试样中分析元素和内标元素的吸光度比Ax/An,根据所测得的吸光度比值从校准曲线求得试样中被测元素含量Cx。




































