1. 方法原理
试样用硫酸+混合催化剂(硫酸钾、硫酸铜)消化溶解后,铵离子在碱性条件下加热蒸馏,生成氨气并与水蒸汽经过冷凝后,被硼酸吸收生成硼酸铵。以溴甲酚绿+甲基红混合溶液为指示剂,用酸标准溶液自动滴定,根据标准酸消耗量可计算出氮、蛋白质的含量。化学反应过程如下:
 (1)消化过程:消化过程为将含氮化合物和浓硫酸共热,将有机氮转化为无机氮硫酸铵:2NH2+H2SO4+2H+ (NH4)2 SO4(其中CuSO4做催化剂)。
(1)消化过程:消化过程为将含氮化合物和浓硫酸共热,将有机氮转化为无机氮硫酸铵:2NH2+H2SO4+2H+ (NH4)2 SO4(其中CuSO4做催化剂)。
(2)蒸馏过程:将消化完的样品转移到定氮仪蒸馏装置,加入过量的浓氢氧化钠,将NH4+转变成NH3,通过蒸馏把NH3驱入过量的硼酸溶液接受瓶内,硼酸接受氨后,形成四硼酸铵。
(NH4)2 SO4+2NaOH=2NH3+2H2O+Na2SO4
2NH3+4H3BO3=(NH4)2B4O7+5H2O。
(3)滴定过程:用标准酸滴定,直到硼酸溶液恢复原来的氢离子浓度
(NH4)2B4O7+2H++5H2O=2NH4++4H3BO3。
2. 主要试剂及溶液
硫酸铵(优级纯),其他试剂为经确认的分析纯试剂,所用溶液按国标规定要求配制及标定。主要试剂如下配制:
40%氢氧化钠溶液:
2%硼酸溶液:
0.1000 mol/L的1/2硫酸标准溶液:
5%溴甲酚绿+1%甲基红混合指示剂(1∶1):
混合催化剂(无水硫酸铜∶硫酸钾=50∶1000):
3. 主要仪器及设备
主要仪器包括:全自动凯氏定氮仪(含300 mL定氮管,硼酸溶液、氢氧化钠溶液、盐酸标准溶液的专用桶);定氮消解炉,万分之一分析天平。
4. 测定方法
样品制备:称取己制备的试样1.0 g(精确±0.1 mg)于消化管中,加入20 mL硫酸和20 g混合催化剂,于石墨消解炉上消化至溶液澄清透明后,定容至100 mL容量瓶中摇匀,分取10.0mL样液至定氮管中,参照仪器操作规程进行分析。
5. 测定条件选择
5.1消化时间的影响
称取同一化肥样品,在不同消化时间下进行样品消化,消化时间对测定结果的影响见表1。
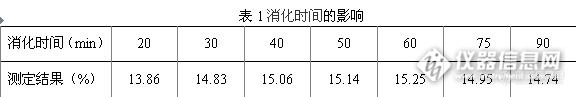
由表1可以发现,消化时间过长会引起氨的损失;消化时间不够,消化不完全也会导致总氮量偏低。根据实验结果,样品消化时间为样品溶液沸腾后40 min ~ 60 min中最好。
5.2催化剂用量的影响
根据标准文献,混合催化剂(硫酸钾、硫酸铜)配比为1000 g∶50 g,但混合催化剂加入量对样品溶液消化有很大的影响,是消化成功的关键,它决定酸的沸点,也决定消化所需的时间。添加硫酸钾可以提高温度加快有机物分解,但硫酸钾量过大,沸点太高,生成的硫酸氢铵就分解放出氨,使氮损失。因此当硫酸过多的被消耗或样品中脂肪含量过高时,要增加硫酸的量。消化时热源的强度同迅速消化和完全氨化关系很大。称取制备好的化肥样品,加入相同的硫酸和不同混合催化剂用量,结果见表2。
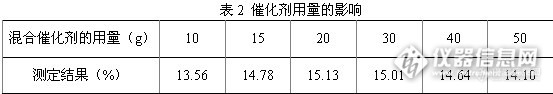
表2结果表明,催化剂加入量对样品溶液消化有很大的影响,当催化剂用量在20 g ~ 30 g时,样品测定结果最高,因此,确定催化剂加入量为20 g。
6. 方法学考察
6.1 线性范围
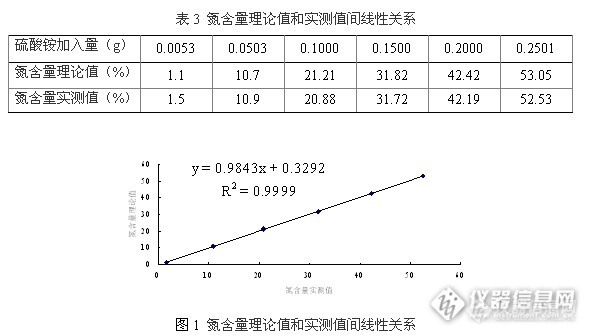
选用纯度>99.9%的硫酸铵,将其在105 ℃的烘箱中烘2 h,然后使用万分之一天平准确称取0.005、0.01、0.05、0.10、0.15、0.20、0.25、0.30 g(精确至±0.0001g)加到含1.00 g的NaCl的10 mL水溶液中,溶解后在凯氏定氮仪上分别测定其氮含量,其氮含量理论值和实测值间线性关系见表3和图1。图1表明,该方法氮测定线性范围为1.1 % ~ 52.53%。如样品测定结果其氮含量大于52.52%,则须稀释后测定。
6.2 最小检出限
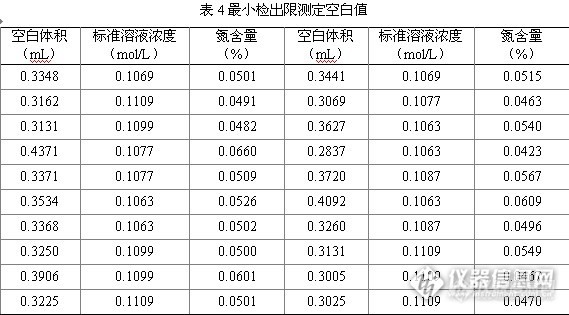
对20次空白值测定结果如表4所示,根据表4计算得,空白值的氮含量标准偏差S为0.0056%,方法最小检出限N含量(%)为4.6×S = 0.026%
6.3 仪器精密度
用纯度>99.9%的硫酸铵,将其在105℃的烘箱中烘2 h,然后使用万分之一天平准确称取0.1g(精确至±0.0001g),并用蒸馏水溶解,放在不同的定氮管中,在凯氏定氮仪上分别测试其氮含量。进样7次。结果见表5
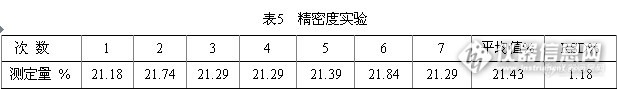
6.4 仪器再现性
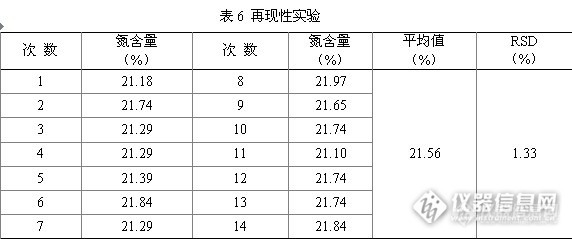
用纯度>99.9%的硫酸铵,将其在105℃的烘箱中烘2 h,然后使用万分之一天平准确称取0.1g(精确至±0.000 1g),并用蒸馏水溶解,放在不同的定氮管中,在凯氏定氮仪上分别测试其氮含量。多次实验结果见表6(不同时间进行的测定)。
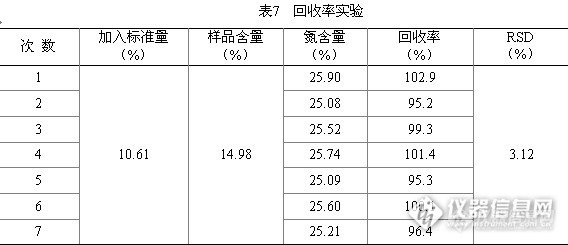
6.5 回收率
称取制备好化肥样品7份按“4”项制备,测得氮的含量结果见表7,实验结果表明回收率在95.2%~102.9%,RSD为3.12%,说明本方法准确度较高。
7. 适用范围
该方法适用于化肥、糕点、肉及肉制品、乳饮料、大豆及其制品、花生、米中氮及蛋白质的测定。根据不同的样品氮换算为蛋白质的系数不同。
糕点、肉制品: 6.25
乳饮料: 6.38
大豆及其制品: 5.71
花生: 5.46
米: 5.95



































