溶剂效应”会导致哪些峰形异常?如何避免溶剂效应?在HPLC分析中样品溶剂的选择和流动相是怎样的联系?为何建议使用流动相溶解样品?。。。。。一系列的问题都和样品溶剂的选择密切相关,如何解决真正的溶剂效应问题,又如何分辨哪些是非溶剂效应问题,不同的问题有不同的解决方法,有哪些前辈留下的实战经验可供分享呢?一起来看看吧!
什么是溶剂效应?
样品溶液的溶剂强度强于流动相溶剂强度时可能会造成的峰展宽、峰分叉现象。即色谱图上较早洗脱的峰前沿或者开叉,与此同时较晚洗脱的峰则较为正常的现象。例如样品溶液的溶剂是100%乙腈(100%的强溶剂),而流动相的组成则较弱,18%的乙腈与72%的水。第一个峰是开叉的,并且与第二个峰相比,明显地变宽了。当样品溶液的溶剂变成流动相时,所有的峰形都改善了,且变得尖锐。
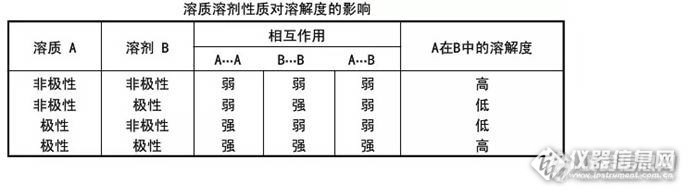
可能发生溶剂效应的情况:
出峰时间早;
保留弱;
进样量大。
用流动相溶解样品能够得到很好的峰形
当溶解样品的溶剂不同于流动相时,样品溶剂与流动相发生混合,样品溶剂被冲稀。如果进样溶剂之强度高于流动相,样品在瞬间会表现为在较强溶剂中,并以较快速度通过色谱柱。表现在色谱图上就是∶色谱峰的保留时间缩短。`当进样溶剂与流动相混合时,一部分分子
会先与流动相混合,致使这些分子通过色谱柱的速度发生变化,使峰形扭曲,发生变形。
洗脱较早的色谱峰峰变形要比洗脱晚的色谱峰严重。解决进样溶剂问题的关键是使进样体积足够小,这样稀释过程会非常快;或使用比流动相弱的溶剂溶解样品。较弱的溶剂会使样品在色谱柱上发生浓缩,在某些情况下,色谱峰会比使用较强溶剂时窄一些。
因此,在通常情况下,如果溶解样品的溶剂比流动相强,进样体积应不高于25mL。进样体积大小与进样溶剂与流动相之间的差别大小有关。
这一差别很容易凭经验得到∶逐渐加大进样体积,直至发生峰变形现象。采用比发生峰变形时小一些的进样体积即可。问题中发生的峰变形就是因为溶解样品的溶剂甲醇强度远远高于流动相,因而得到了变形的、展宽的峰。
如果使用水来溶解样品,溶解样品的溶剂弱于流动相,峰形会得以改善。在离子对色谱中,我们建议使用流动相来溶解样品,以最大程度地降低基线假峰的出现。
样品溶剂分别为100%乙腈、流动相时峰形分析
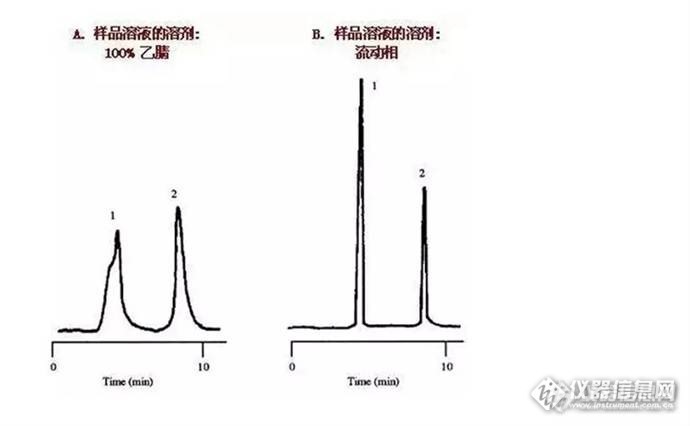
上图中,色谱图上较早洗脱的峰扭曲变形或者开叉,与此同时较晚洗脱的峰则较为尖锐与对称,这些现象显示一个比较特殊的起因——样品溶液的溶剂很可能强于流动相。此种强溶剂效应的例子在左图中可见。此处的样品溶液的溶剂是100%乙腈(100%的强溶剂),而流动相的组成则较弱,18%的乙腈与72%的水。第一个峰是开叉的,并且与第二个峰相比,明显地变宽了。当样品溶液的溶剂变成流动相时,所有的峰形都改善了,且变得尖锐。见右图。
为什么呢?这是因为当样品进样时,有可能出现峰展宽,最佳的样品溶液组成和体积将会保持在10%甚至更低,在这个例子里,当样品溶剂与流动相溶剂强度不同时,换句话来说,也就是样品未用流动相溶解,因此,有些样品分子溶解在强溶剂中,并随强溶剂流过柱子,而有些则溶解在流动相中,从而导致峰分叉.
当样品与流动相强度相差较小,进样影响也会小,第一个峰可能会宽于第二个峰,而当这种展宽导致必要的分离度降低时,这样情况应引起注意,在左图中,使用一根短柱,和5μL进样,这与最佳进样体积4μL相近,用了极性更强的溶剂导致分离度明显的降低,从2.1降到1.5(如右图,分离度为2或更大是评估一个完善方法的一个必要参数,也是每天方法的验证参数,1.5只是一个基本的分离度,任何一个方法或一根柱子都必需满足这个条件,当进样为一倍时,也就是10μL时,分离度更一步降低,此方法就不行了。
样品溶剂最好具有哪些特点比较好?

实战问答Q&A,绝对接地气!
1.HPLC里面的溶剂效应是如何产生的,该如何避免?
如何判断分叉的峰是否由溶剂效应造成,第一就是通过HPLC仪器比对一下两个劈叉峰的紫外吸收波谱,看看他们是否能完全一样的μV特征吸收,第二将进样体积调节到0.1μl看看,峰是否好转,如果满足上面两个条件几乎就可以肯定是100%的溶剂效应。
产生原因1,样品溶液的溶剂很可能强于流动相。例如样品溶液的溶剂是100%乙腈(100%的强溶剂),而流动相的组成则较弱,18%的乙腈与72%的水
产生原因2,就是进样量比较大,
产生原因3,样品的pH值与HPLC流动相pH悬殊,比如酸性流动相体系,样品pH偏碱性(pH=10),这种情况下也很有可能发生其实上面三个原因归结起来就是一个原因,那就是样品的溶剂与流动相存在一些不一样,当样品进入流动相的时候,由于这种巨大的差异,一部分样品溶解进入了流动相,一部分还留在进样的溶剂里面,所以在HPLC上出现了保留的差异,所以要解决这个问题的最佳方案就是,使用流动相的起始梯度溶解样品,如果由于溶解度的问题,不得不改用其余的溶剂,那就只能降低进样体积来解决,比如 5μl,1μL。
2.高效液相做专属性实验时,溶剂峰和目标峰出峰时间一样怎么办?分离的是氨基酸类物质,不是离子型化合物,流动相用的甲醇和水,也是用流动相配的溶剂,但是比例有少许差别,单进溶剂时确实有溶剂峰,用流动相配的样品可以和溶剂峰分开,我在做肠灌流实验,但是当样品进入动物体内灌流后,进样后就分不开了,我想把样品的出峰时间延长一些,认为能分开,但是不知道怎么调整,实在没办法。
回答:
问题1:你用的稀释剂是纯的有机相吗?稀释剂中有机相的比例一般尽可能与流动相一致,以减小溶剂效应.如果改变稀释剂仍不能改善,则说明你的色谱条件不合适,检测组分在色谱柱中几乎无保留.如果你的化合物是离子型的,一般需要使用缓冲盐,缓冲液pH值应在化合物pKa值正负2之外,以增强组分在色谱系统中的保留.如果不是离子型化合物,则减少有机相比例,以增加组分保留时间,避开溶剂峰.
问题2:氨基酸类物质是两性化合物,出峰时间跟pH值有很大关系,样品经过动物体内后,pH值肯定是发生了变化的,样品的预处理就很重要了(这个你要请教其他高人,我没做过生物方面的药物分析)。如果你只是想单独延迟样品出峰时间,可以试着减少有机相比例,看能不能达到效果,如果不行,可能还是要用缓冲体系。
3.醋酸曲安耐德用高效液相色谱测定,流动相为甲醇-水(60:40),ODS C18柱,紫外检测波长240nm,柱温30度。以前样品用甲醇溶解后直接进样,色谱峰各项参数正常。现在用甲醇溶解后进样,色谱峰变宽,必须用流动相溶解才能正常。仪器与色谱柱未更换,溶解样品的甲醇换成默克色谱纯甲醇也还是这样!什么原因?另外我需要测定的另一个中药中黄芩苷也出现了这种现象?我现在该怎么办?
解答:溶剂效应是肯定的。溶剂效应的原理其实很简单:就是溶质的极性和流动相的极性不匹配,导致溶质被流动相包裹或部分包裹,不能充分分散并充分与C18交换,所以才导致峰展宽。你的醋酸曲安耐德极性并不是很强。我帮你查了它的log P,没查到,但根据我的经验,并根据其结构式我大概推算了一下,应该在2左右,应该属于中等偏弱极性的化合物(C26)。所以其在60:40这样含水流动相匹配度肯定不好。以前你做的没有问题,一个原因是上面各位所说的,溶解样品的甲醇可能含水量稍微高一些,比如大约含水2%(只是猜测哈),可能就不会出现溶剂效应,而这次用的甲醇含水量很低。另一个可能的原因我觉得可能是原来柱子相对比较新,用了一段时间后,柱头可能有塌陷,导致溶剂效应加剧(这种可能性更大一点)。这些都是猜测的,否则很难解释以前很好,现在不好的原因。
无非就是这两条。至于黄芩苷,我也查了其结构式,极性跟醋酸曲安耐德也差不多,能稍微强一点。同样的问题。解决办法:你可以尝试用90:10的溶液溶解样品再试一下,如果变好了,那就说明溶剂效应很明显。只能改变溶剂样品的溶剂了,没别的办法。
关于消除溶剂效应,在我们使用的甲醇当中,其实并不是单单甲醇,还有其他成分,只是这些个成分,在紫外下不干扰测定而已做过分离的朋友都知道,不同厂家,不同批次间甲醇的洗脱能力,经验证明确实不尽相同的,特别是对于对极性条件相对苛刻的分离情况。所以,问题可能在于甲醇上,有条件的话,是否可以测一下这个批次甲醇的水分呢。当然,溶剂效应还有其他方面的影响,比如系统,色谱柱...
4.今天我做葛根素的测定,葛根素是用30%乙醇溶解,流动相是甲醇-水(25:75),半年前的色谱峰对称系数为1.1,现在变成了1.45,峰明显变胖!为什么现在只要不是用流动相溶解,就会出现峰变形的情况呢?与我的仪器是否有关系,还是溶剂(比如甲醇)有关呢?
一、葛根素和黄芩苷包括醋酸曲安耐德他们都是结构非常相似的三个化合物,所以一个出问题,所有的都会有相似的问题的。我觉得问题最大的可能还是在色谱柱的柱头。溶剂效应现象,柱子越细,进样量越大,目标化合物和流动相极性匹配度差异越大,问题越明显。如果你用的是超高压的话,会更明显。我在做HPLC/MS/MS的时候,用的都是2.1mm的柱子,基本上都要用流动相来溶解样品,否则绝大多数情况下都有溶剂效应。你如果用超高压HPLC这种溶解效应会很显著。这种问题没别的办法,只有改用流动相溶解。
二、你上面提到的葛根素的测定要做一个对比实验:一个是纯溶剂溶解的,一个是流动相溶解的。如果两者有区别,那说明是溶剂效应问题,建议先用异丙醇清洗系统,如果溶剂效应还在,建议再换成20微升进样定量环,每次进10微升。满环进样还是有很明显的溶剂效应,工程师也没有跟好的办法。
如果二者没有区别,那就不是溶剂效应问题。你半年前做的好好的,不代表这根柱子现在就必须一定得好好的,如果柱子使用时间长了,脏了,对称因子从1.1-1.45也是非常正常的。我不知道你做过对比实验没有?
如果单纯是拖尾的问题,那很简单,你把柱子入口部分打开,将柱头脏的部分用一个特制的小铲取出来,再用C18填料(散装,最好是进口的,不限品牌)用乙腈调成糨糊状,将缺少填料部位填实(注意填料别弄的到处都是,影响密封效果),再装好,重新使用,就会恢复到以前的1.1的状态。如果塌陷的比较多,填完正常使用一两次,让高压将后填的填料压实,再重复填装一次。这样这根柱子还会继续使用很长时间。柱子柱效降低或者峰拖尾90%都是这个原因。



































