气相色谱(GC)
先来看看气相色谱的气路与温控系统
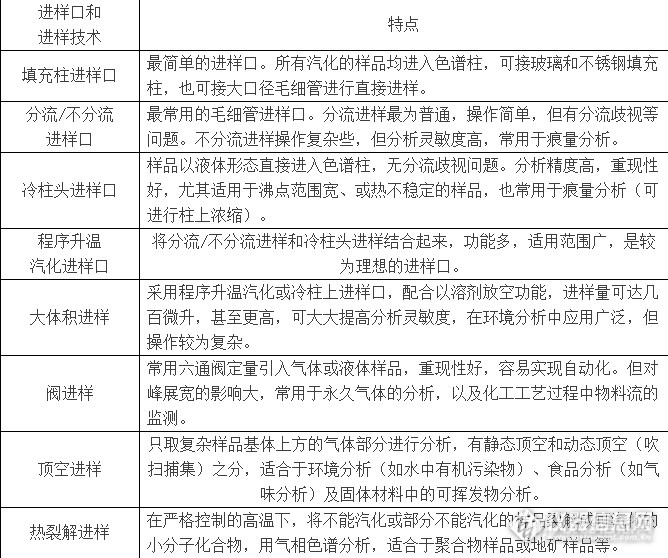
再来看看气相色谱的进样系统
气相色谱最重要的分离系统
检测系统会出现哪些问题?
仪休哥再说说气相色谱的信号记录系统
zyl3367898
第1楼2016/03/23
基础知识,也是必须知道的知识。
liandong
第2楼2016/03/23
总结的太好了。
symmacros
第3楼2016/03/24
非常全面的气相色谱资料,内容很棒。
PAEs
第4楼2016/03/25
感谢楼主的分享,好东东!
第5楼2016/03/28
是《气相色谱百问精编》里的内容。
xdzh130
第6楼2016/04/02
谢谢分享!
第7楼2016/04/02
其实许多做色谱的人也是不完全清楚所以这些方面的,还是要多学习一下。
Frostmoure
第8楼2018/04/03
66666
dadgoh
第9楼2018/04/03
干色谱这行就是不断积累经验的过程。
01524101
第13楼2018/04/04
学习
品牌合作伙伴
执行举报