

皮皮鱼
第5楼2017/08/30
这个主题是比较深入的,很多朋友从来没考虑过,而且没有一定基础还不能理解。
更大的问题是:理解了之后,要改变目前的工作习惯,更难。
首先是用一瓶不是控制指标的标准气标定仪器,自己心里就容易没自信了。
其次是和别人比对的时候,别人一看你用的标准气不是控制指标,直接就敢指责你的标准气不合理,然后判定你的数据不可靠。而你想和他说清楚这些道理蛮难的。
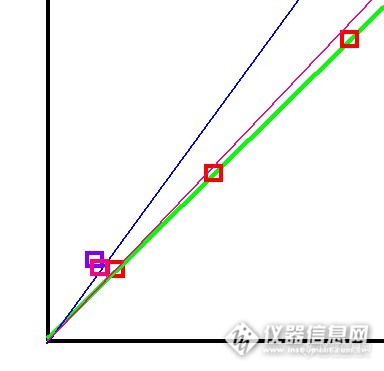
但无论从理论上,还是事实上,都是用高浓度标准气可靠的多。我在做技术员的时候,从来不会定10ppm以下浓度的标准气,一般至少定20ppm以上。而且在数据争议的时候从来没有输过。。。哪怕控制指标是1ppm,我也会定制20ppm的标准气。
但改变朋友们的习惯真的很难。所以我现在都建议朋友定制两瓶标准气了,一瓶控制指标的,一瓶高一些浓度的。有对比才会有结论,实践是检验真理的唯一标准。两瓶标准气互相标定,一起使用,慢慢就会了解单独用哪一瓶更有可靠了。
习惯是慢慢改过来的。以前朋友们液相进样,手动注射器,都是推针后就马上拔出来,现在已经有很多朋友停留2秒再拔出来了,因为她们都自己实际测试过效果了。标准气的事情,要实际检验更难做到一点,但也不是完全做不到,慢慢的来说,慢慢的改变吧!


































