

表1仪器条件
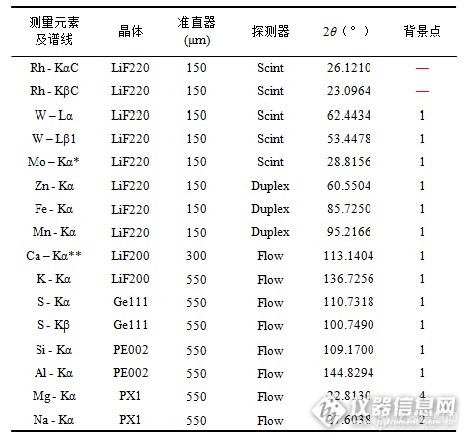
*选择黄铜滤光片。
**表中Ca以前元素测量电压/电流为60 kV/60 mA,Ca及Ca以后元素为30 kV/120 mA.
1.2 实验方法
先行称取~2 g熔剂铺垫于铂黄坩埚底部,而后称取~4 g熔剂于100 mL瓷坩埚内,加入样品(记录称样量,取样量为:原矿、尾矿1g至3 g,中矿、精矿0.35 g至0.80 g)及3 g NH4NO3,混合均匀,转移至铂黄坩埚内,补加熔剂均匀覆盖表面。于600℃预氧化30 min,随后升温至1100 ℃(为保证低稀释比熔融,故未选择低温条件),熔融 5 min,加入NH4I,摆动5 min,静置60 s后出炉,冷却后记录样片质量。
2结果与讨论
2.1预氧化程序
硫化物精矿还原性较强,若预氧化不完全,在熔融过程中可能腐蚀铂黄坩埚。为保证预氧化效果,设计验证试验如下:以标准序列中S含量最高的样品GBW07144(取样量0.5000 g,NH4NO32g,n=3)为实验对象,样品准备流程参照1.3,坩埚(含物料)入炉前称重,记为m1,对照空白坩埚称重为m1b,预氧化30 min后取出,称重分别为m2和m2b,计算得样品灼烧变量((m2-m1)-(m2b-m1b)),仅为0.16xxg,与理论值(GBW07144中Mo、S配分与辉钼矿MoS2相近,其差异可以解释为样品中部分S源于硫铁矿或单质硫。假设样品中S均以硫化物或单质硫形式存在,在充分预氧化的情况下,Mo、S氧化产物依次为MoO3与SO3,0.5 GBW07144的理论灼烧变量约为0.3735 g)存在显著差异,遂将预氧化时间延长至1 h,灼烧变量增至0.29xxg。比较数据发现相比对照空白,由于发生氧化还原反应,样品中NH4NO3消耗速度更快,在30 min内已消耗殆尽,将预氧化时间延长至1 h其质量并无变化,而对照空白中的NH4NO3则在约40 min后方才分解殆尽,以上情况解释了灼烧变量的增加。为探索预氧化效率能否进一步提高,NH4NO3用量依次增至3.0、4.0 g(因硝酸铵分解可能产生多种氮氧化物,根据化学反应理论计算确定其用量的方式不切实际),结果发现样品灼烧变量维持不变,即0.29xx g为通过以上预氧化程序可获得的灼烧变量上限。与此同时,我们发现根据样片质量计算所得的灼烧变量亦小于理论值,约为0.32xx g(灼烧变量=样片质量-对照空白质量-称样量),但需注意的是,样品中各待测元素对硼酸锂盐熔剂挥发的影响及样品组分的挥发均难以准确量化。因此,在通过上述验证实验判断熔融制样预氧化程序是否可行时不应依赖理论计算(因缺乏对样品中各元素赋存状态的详细研究,理论计算值必然存在偏差),在预设温度条件下,若氧化剂用量不同而样品灼烧变量基本保持恒定,即可认为预氧化程序安全有效。在此基础上本实验探索了在更低温度条件下以更低稀释比预氧化精矿样品的可行性,预氧化温度设为400 ℃,取样量增至0.8000 g,结果发现400 ℃条件下对照空白中NH4NO3分解缓慢,即便将预氧化时间延长至1 h以上,仍有残留,且质量基本保持恒定,导致计算所得的样品灼烧变量偏低,需在30 min后升至600 ℃加速其分解,以判断与氧化效果,结果表明NH4NO3用量依次为3.0、4.0、5.0 g的样品灼烧变量基本一致,证明在400 ℃条件下以3.0 g NH4NO3在30 min内可氧化0.8000 g钼精矿样品,但在本实验中进一步降低精矿稀释比作用并不明显,因此工作曲线中精矿、中矿等稀释比仍维持在约16:1。此外,在处理含铅锌等硫化物的重晶石矿物时,因硫化物可能为重晶石晶体包裹,应采用较高的预氧化温度。
熔融制样定量分析的基础在于待测元素在样片中分布均匀,砷、锑、铋、碲等元素氧化物易挥发,在熔融制样过程中亦无法避免,在变稀释比熔融制样方法中,熔体表面积、流动性等可能影响挥发的因素相比均一稀释比方法差异更大,对上述元素工作曲线线性及分析结果的准确度、精密度影响亦相应增加,在进行基体效应校正及结果分析时应注意。
2.2工作曲线
钼矿石标准物质数量稀少,且含量跨度巨大,需加入人工混标。在标准配制过程中采用如下两种方法:1.参考文献,以GBW07141与GBW07144为基础,通过改变稀释比(80~2.667:1)及两者间配比(w7141:w7144=9~1:1)的方式建立工作曲线;2. 将GBW07141-7144与土壤、沉积物、岩石等混合,以解决中方法1中造岩元素配比单一的问题。在输入标准序列浓度时试验了两种模式:1. 样品类型选择熔片,稀释比设为可变,逐一输入称样量及样片质量;2. 样品类型选择固体或粉末压片,标准中各待测元素含量按下列公式计算后录入(即待测元素在样片中的质量浓度)。

各工作曲线标准序列及浓度模式选择见下:
表2 工作曲线
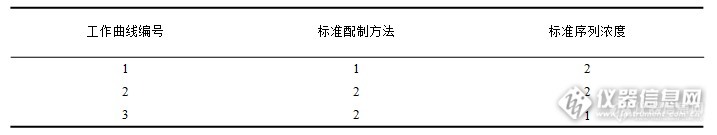
以康普顿散射内标法校正Mo、Zn、W,但造岩元素以基本参数法或理论α系数法校正均无法获得满意的工作曲线,只能选择经验系数法,其曲线外推效果较差的缺点可通过在标准中添加对应元素的方法解决,而可能出现的过度校正问题则需要以一系列不同含量水平的实际样品加以验证。1号曲线因样品来源单一,元素配分缺乏变化,对实际样品中造岩元素分析效果欠佳;2、3号工作曲线改进了上述情况,3号工作曲线相对简单直观,2号曲线的优点则在于其受灼烧变量、熔剂挥发等因素的影响更小。本实验选择3号曲线开展后续研究。
与此同时,某些中矿样品因分离过程尚未完成,干扰元素含量可能远超标准预设值,其影响可能被忽略,将导致分析结果异常。为解决上述问题,可考虑在部分标准中补充以上元素并复熔,本实验中W即为后续补充。
2.3准确度、精密度与检出限
以实际原矿、中矿及精矿样品(在变稀释比条件下)开展精密度研究,精密度较差,未满足《DZ/T 0130.3-2006 地质矿产实验室测试质量管理规范》中标准偏差值要求,后以标准物质GBW07141、7144代替,结果有明显改善,见下表。
表3 精密度实验

通过SuperQ软件计算得到的检出限随稀释比波动,即便参考方法建立模式二,将检出限定义为待测元素在样片中的质量浓度,但样品间基体差异的问题仍无法解决,故在讨论检出限时,应指定适用范围(如尾矿样品中Mo的检出限、精矿样品中SiO2的检出限),以上检出限数据亦更具实用价值。
因实际样品的均匀性问题,故部分元素方法对照结果存在差异,但其中主量组分及Mo相对误差较低,相对误差较高的次量分析结果亦具有一定参考价值,同时,本方法重现性良好,因未固定取样量及熔剂用量,在称量时间方面亦有优势,适用于日常分析。与其他方法对照时CaO、MgO、K2O的测定采用焙烧-四酸溶解-ICP-OES分析,Fe、Mo、SiO2的测定采用过氧化钠熔融-硝酸提取-ICP-OES分析,S的测定则采用燃烧中和法。结果见表4:
表4 方法对照实验

3 结论
本实验通过变稀释比熔融建立了适用于钼矿石选矿流程样品中主量至微量元素分析的X射线荧光光谱分析方法,在一定程度上克服了单一稀释比方法检出限不足的问题,提出了一套合理可靠的预氧化程序验证方法,指出判断预氧化效果无需依赖理论计算,当预氧化温度在400~600 ℃,使用NH4NO3做为氧化剂,有效预氧化时间不超过30 min。在配制标准序列过程中各待测元素浓度应呈梯度,配比多样。以康普顿散射内标法校正Mo、Zn、W,以经验系数法校正造岩元素。方法准确有效,重现性良好,变稀释比熔融方法亦可应用于锡矿石、铬铁矿的分析。
本实验的灵感来源于Adnan Younis, ZohrabAhmadi, Matthew G. Adams, Amir Iqbal. X-Ray Spectrometry, 2017, 46(1): 69~76。
不足之处敬请指出,多多讨论~
玉米馒头
第5楼2017/11/15
1. 条件没反,轻元素选择低电压高电流分析;
2. 表3的精密度是平行样的分析结果,倘若是固定稀释比,就XRF而言精密度并没有太多值得研究的地方,正因为是变稀释比所以想看看;
3. 表3中没有单位,表4中单位均为%,忽略了;
4. 重点一则是介绍变稀释比方法,因为参考文献中应用变稀释比方法处理的样品相对简单,而硫化物及锡矿石等难溶样品更适用于这类方法的应用;二是为熔融前的预氧化时间设置提供一些参考。就我个人而言,现阶段对于传统格式的文章并不是太感兴趣,在结果与讨论部分放进一堆有的没的参数实验,其可推广性存疑,所以我更倾向于授人以渔,为读者提供一个可供参考的思路;但这样一来文章的体量就单薄了,所以说一是得尽量做得细些,二是多加入些我自以为的“干货”
5. 方法对照实验算是偷懒了,因为化学分析这块挺久没接触了,所以只用了客户送检的项目结果。

































