UPLC-MS/MS法测定黄芪水提物中的黄芪甲苷含量
传统中药的煎煮,大多是用水作为溶剂,因此研究药材的水提物中有效成分的含量对药材的工业生产和实际应用具有不可忽视的指导意义。据文献报道,黄芪水提物中主要为糖类和皂苷类成分,而水提物药渣中则是提取后剩余的淀粉、纤维素、木质素等原药材的基体成分和一些不易溶于水的酯类、酮类和芳香类成分。黄芪甲苷作为黄芪皂苷中一类重要的物质,已被药典收录为黄芪含量测定的指标成分。为了保证临床应用制剂的质量和进一步开发利用中药黄芪,研究黄芪水提物中黄芪甲苷的含量显得格外重要。在预实验中,笔者发现药典中测定黄芪中黄芪甲苷含量的方法并不完全适用于黄芪水提物中黄芪甲苷含量的测定,预处理时出现乳化现象导致萃取不完全,另外利用药典中规定的蒸发光检测器液相色谱仪测定无法得到理想的线性方程。因此,笔者结合黄芪水提物的性质和仪器灵敏性等特点,利用UPLC-MS/MS法考察黄芪水提物中的黄芪甲苷含量测定的方法学和并利用该方法测定黄芪水提物中黄芪甲苷的含量。
1 材料和仪器
1.1 样品
收集9组黄芪水提取物样品,黄芪饮片为济南济成堂中药饮片有限公司提供(批号18033101)。
1.2 试剂
黄芪甲苷对照品(成都瑞芬思生物科技有限公司批号H-013-170117),乙腈为色谱纯(天津市科密欧化学试剂有限公司);超纯水。
1.3 仪器
Waters XevoTQ-S三重四级杆质谱仪(美国Waters公司)。ACQUITY UPLC H-CLass超高效液相色谱仪(美国Waters公司);超声波清洗机KS-300E(宁波科生仪器厂);电子天平MS205DU(梅特勒/瑞士)。
2 方法学考察
2.1 色谱与质谱条件
色谱条件:色谱柱为ACQUITY UPLC BEH C18柱(2.1*50 mm,1.7 mm)。流动相为乙腈-水(A∶B)系统。梯度洗脱,A相:0→1 min A保持30%;1→2 min A为30→60%;2→3 min A为60→70%;3→4 min A保持70%;4→4.5 min A为70→30%;A相:4.5→5.5 min A保持30%。流速 0. 2 mL/min,柱温为40 ℃,进样体积 1. 0 μL。
质谱条件:电喷雾正离子检测模式,毛细管电压;3.0 kV;脱溶剂气流:N2,流速800 L· h-1,脱溶剂温度400 ℃;锥孔电压:40 V,锥孔气流:N2,流速150 L·h-1;离子源温度: 400 ℃;雾化气压力为7.0 bar碰撞气体: 氩气。采用MRM 定量模式,质量扫描范围为100~1 000 amu。该条件下,获得黄芪甲苷的分子离子峰 m /z 785. 42,子离子峰m /z 143.08。黄芪甲苷提取总离子流图和一级质谱图分别见图3-1和图3-2。
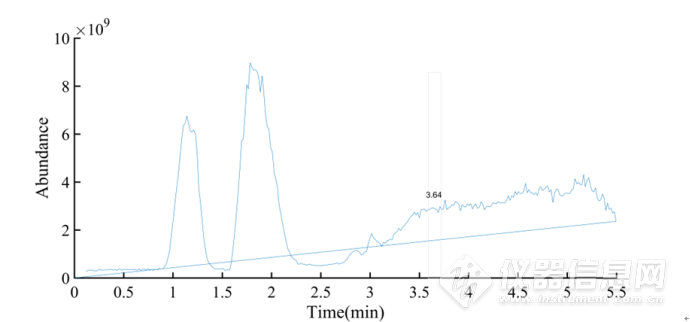
图3-1 黄芪甲苷的总离子流图
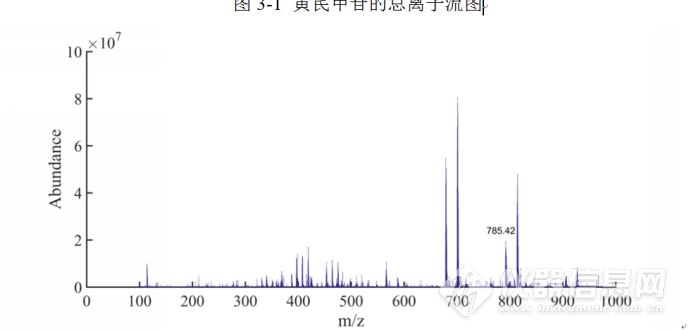
图3-2 黄芪甲苷的一级质谱图
2.2 供试品溶液的制备
精密称定1/200重量的黄芪水提物样品(折合黄芪药材0.5g)置于锥形瓶中,精密加入4%氨水溶液100 mL,称定重量,超声30 min,取出放至室温,用4%氨水溶液补足减失的重量,摇匀,过滤,精密吸取续滤液10 mL过Dikma ProElut C18-U-SPE柱(先以甲醇20 mL活化,再以水20 mL平衡),上样后用10 mL水淋洗,弃去,再用适量甲醇洗脱,收集洗脱液,定容至10 mL容量瓶中,摇匀,用0.22 μm的微孔滤膜过滤,即得。
2.3 对照品储备溶液的制备
精密称取黄芪甲苷标品1.01mg至10 mL容量瓶中,用甲醇溶解后定容,吸取1 mL至100 mL容量瓶中,用甲醇定容,制成浓度为1.01 μg/mL的对照品储备溶液。
3 结果
3.1 线性关系考察
精密吸取 2. 3 项下对照品储备溶液 1,3,5 mL,用甲醇稀释并定容到 10mL 容量瓶中,制备0.1,0.3,0.5 mg·mL-1质量浓度对照品溶液。另吸取0.1,0.3 mg/mL质量浓度对照品溶液5 mL至10 mL容量瓶,用甲醇定容,得到0.05,0.15 mg/mL质量浓度对照品溶液。在拟定分析条件下,准确吸取 10. 0 μL 进样分析。黄芪甲苷离子选择 m/z 785. 42 为母离子,143.08 为子离子,准确吸取1 0 μL 进样分析。以质量浓度为横坐标,以提取离子流图峰面积为纵坐标进行线性回归,如图3,得回归方程为:Y=46.135X + 1938.8(r2= 0. 9984),提示黄芪甲苷在0. 05~0.5 mg/m L 范围内线性关系良好。黄芪甲苷对照品标准曲线如图3-3所示。
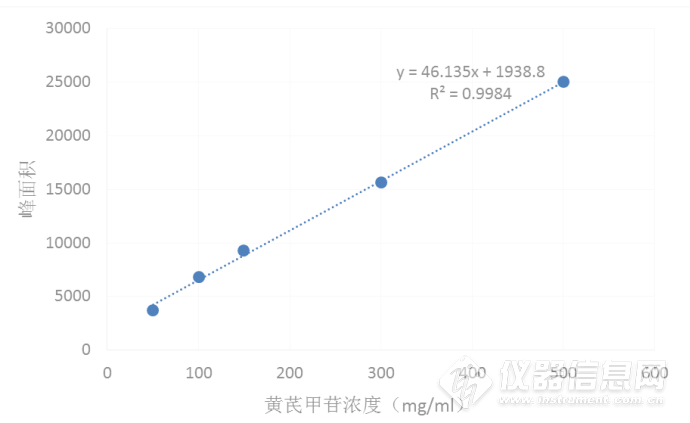
图3-3 黄芪甲苷标准曲线
3.2 精密度实验
在拟定分析条件下,精密吸取供试品溶液1.0 μL,连续进样 6 次,记录提取离子流图峰面积,测定黄芪甲苷量,计算得相对标准偏差RSD为1.1% ,提示该方法具有较好的精密度。
3.3 重复性实验
取同一黄芪样品 6份,按2. 2 项下方法制备供试品溶液,在拟定分析条件下,准确吸取 1. 0 μL 进样分析,测定黄芪甲苷量,计算得 RSD为2.0%,提示该方法重复性良好。
3.4 稳定性实验
取黄芪药材供试品溶液,分别在0 h、3 h、6 h、9 h、24 h、48 h后,在拟定分析条件下,准确吸取 1. 0 μL 进样分析,测定黄芪甲苷量,计算得 RSD 为 4.7% ,提示黄芪供试品溶液稳定性较差。
3.5 加样回收率实验
精密称取 6 份黄芪甲苷量已知的黄芪水提物样品,每份折合黄芪药材 0. 5 g,分别准确加入浓度为 0. 0 342 mg /mL 黄芪甲苷溶液1 ml,1 ml,2 ml,2 ml,3 ml,3 ml,按 “2. 2 ”项下方法制备供试品溶液,准确吸取 1. 0 μL 进样分析,测定黄芪甲苷量,计算回收率,结果见表3-1。由表3-1可见,方法平均回收率为 102.35% ,表明该方法具有较好的回收率。
表3-1 黄芪甲苷加样回收率测定结果
| 样号 | 样品中的量/mg | 加入量/mg | 测得量/mg | 回收率/% | 平均值/% | RSD/% |
| 1 | 0.069 | 0.034 | 0.1 | 101.68 | 102.35 | 2.8 |
| 2 | 0.069 | 0.034 | 0.11 | 104.44 |
| 3 | 0.069 | 0.068 | 0.14 | 104.17 |
| 4 | 0.069 | 0.068 | 0.13 | 96.42 |
| 5 | 0.069 | 0.10 | 0.17 | 102.78 |
| 6 | 0.069 | 0.10 | 0.18 | 104.63 |
3.6 黄芪甲苷的含量测定结果
取9组黄芪水提物按照“2.2”项下操作,制备供试品溶液,准确吸取 1. 0 μL 进样分析,测定黄芪甲苷的含量。黄芪甲苷对照品和黄芪水提物样品的MRM 离子流图见图3-4、图3-5。供试品黄芪甲苷含量测定结果见表3-2。
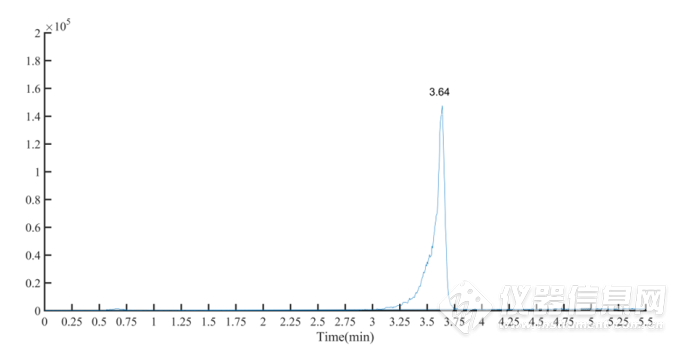
图3-4 黄芪甲苷对照品的 MRM 离子流图
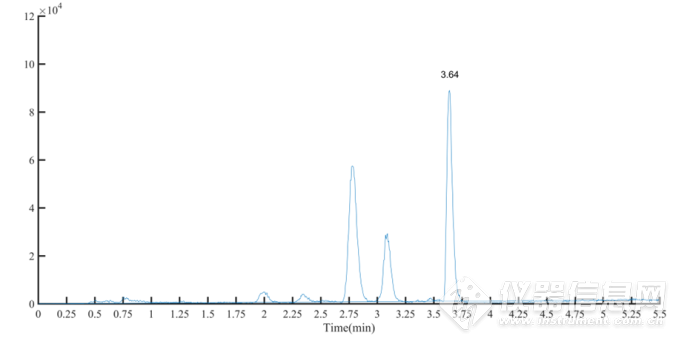
图3-5 黄芪水提物样品的 MRM 离子流图
表3-2 9组黄芪水提物中黄芪甲苷含量测定结果
| 试验号 | 峰面积 | 含量(mg/g) |
| 1 | 5120.00 | 0.14 |
| 2 | 6744.00 | 0.21 |
| 3 | 10275.67 | 0.36 |
| 4 | 26955.00 | 1.08 |
| 5 | 10599.00 | 0.38 |
| 6 | 7212.33 | 0.23 |
| 7 | 11247.33 | 0.40 |
| 8 | 8008.33 | 0.26 |
| 9 | 9171.00 | 0.31 |
3.7 正交试验设计及结果
水提的黄芪甲苷转移率考察正交试验与水提的出膏率考察正交试验设计相同,即以水作为提取溶剂,把影响药材提取效果的用水量(A)、提取时间(B)、提取次数(C)确定为考察因素,以上三个考查因素各分3个水平考察,见表3-3。
表3-3水提实验因素水平表
| 水平 | 因素 |
| A(用水量/倍) | B(提取时间/h) | C(提取次数/次) |
| 1 | 4 | 1 | 1 |
| 2 | 8 | 2 | 2 |
| 3 | 10 | 3 | 3 |
黄芪甲苷转移率=各实验组黄芪提取物中黄芪甲苷含量/原黄芪药材中黄芪甲苷含量×100%。原药材中毛蕊异黄酮苷的含量按照药典的方法测得的结果为0.4042mg/g。跟据实验数据,得到水提实验中设定的不同工艺条件下的毛蕊异黄酮苷的转移率,其中因素D为误差项,作直观分析表和方差分析表,见表3-4,表3-5。
表3-4 黄芪甲苷转移率考察L9(34)正交试验表
| 试验号 | A | B | C | D | 黄芪甲苷
转移率/% |
| 1 | 1 | 1 | 1 | 1 | 34.12 |
| 2 | 1 | 2 | 2 | 2 | 51.54 |
| 3 | 1 | 3 | 3 | 3 | 89.41 |
| 4 | 2 | 1 | 2 | 3 | 268.30 |
| 5 | 2 | 2 | 3 | 1 | 92.88 |
| 6 | 2 | 3 | 1 | 2 | 56.56 |
| 7 | 3 | 1 | 3 | 2 | 99.83 |
| 试验号 | A | B | C | D | 黄芪甲苷
转移率/% |
| 8 | 3 | 2 | 1 | 3 | 65.10 |
| 9 | 3 | 3 | 2 | 1 | 77.56 |
| K1 | 175.07 | 402.25 | 155.78 | 204.56 | |
| K2 | 417.74 | 144.42 | 397.40 | 207.93 | |
| K3 | 242.49 | 223.53 | 282.12 | 422.81 | |
| 优水平 | 2 | 1 | 2 | 3 | |
| R | 242.67 | 257.83 | 241.62 | 218.25 | |
表3-5 黄芪甲苷转移率考察方差分析结果
| 方差来源 | 离差平方和 | 自由度 | F | 显著性 |
| A | 10460.75 | 2 | 1.13 | - |
| B | 7698.00 | 2 | 0.75 | - |
| C | 9736.83 | 2 | 1.02 | - |
| D | 10424.20 | 2 | - | - |
注:F0.1(2,2)=9,F0.05(2,2)=19,*为有显著性,-为无显著性。
从正交试验结果可知,水提实验中,各因素对黄芪甲苷转移率的影响大小顺序为:A(用水量)>C(提取次数)>B(提取时间);每个因素3水平之间的趋势为A2>A3>A1,B1>B3>B2,C2>C3>C1,直观分析得最佳提取工艺为A2B1C2,即加水8倍量,提取2次,每次1h。表3-5的方差分析结果表明: A、B、C三因素对黄芪甲苷转移率的影响都无统计学差异(P<0.05)。
4 讨论
本章实验利用超高效液相串联三重四级杆质谱仪测定黄芪水提物中黄芪甲苷的含量,相比药典中测定黄芪药材中黄芪甲苷的方法,此方法预处理步骤更少,用时更短,目标峰与其他相邻峰的分离度也更大,适合于黄芪水提物中黄芪甲苷含量的测定。但美中不足的是,供试品的预处理需要过SPE柱,仪器的使用和维护费用更高,实验者在测定样品的含量前需要综合考虑。
从测定结果来看,设置的三个因素中,用水量对黄芪甲苷的转移率影响最大,但仍无统计学差异(P<0.05),说明用水量、提取次数、提取时间三种工艺的改变对黄芪水提物中黄芪甲苷的含量无显著性影响。第4、5、7组的黄芪甲苷转移率相比其他组的更高,尤其是第4组测得的黄芪甲苷的含量远高于用药典方法测得的药材中黄芪甲苷的含量,正交实验分析结果也说明加水8倍量,提取2次,每次1h是最佳提取工艺。这一结果与水提实验中第3、4、7组测得的出膏率较高有些差异,第4组实验的出膏率也不算低,但不是最高,而出膏率最高的第3组黄芪甲苷的转移率却不高,这说明出膏率与黄芪水提物中黄芪甲苷的转移率并无明显的对应关系。
合乎标准要求的有效成分含量是保证药物疗效稳定可靠的硬性指标,实现有效成分提取的最大化是优化工艺条件的重要目标。本章关于不同提取工艺条件下黄芪甲苷转移率的考察研究,对芪龙胶囊和黄芪配方颗粒工艺的优化具有指导性意义。
参考文献
黄冬兰,徐永群,陈小康. 黄芪药材及其水提物的红外光谱分析. 光谱实验室,2012,29(5):2823-2826.



































