附件:
iangie
第3楼2021/05/24
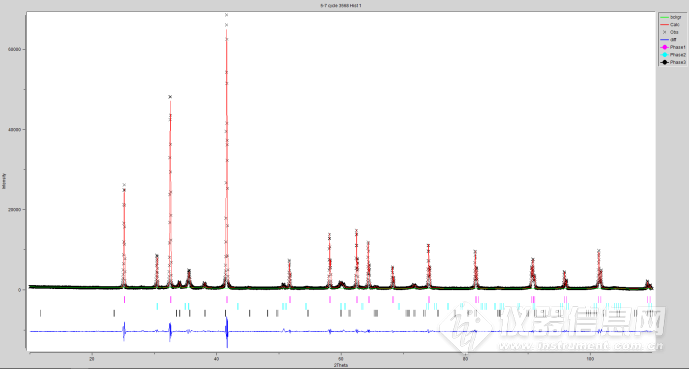
差不多了 TOPAS修出来的Rwp也就7.95 GOF=2.6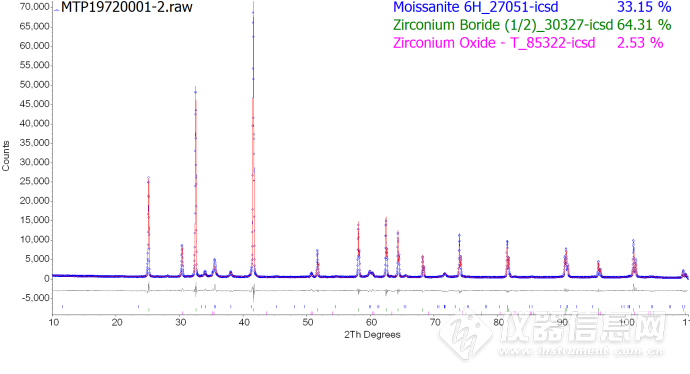
File 1 : C:\VHD\1\MTP19720001-2.raw
Range Number : 1
R-Values
Rexp : 3.06 Rwp : 7.95 Rp : 5.88 GOF : 2.60
Rexp`: 4.40 Rwp`: 11.42 Rp` : 10.17 DW : 0.43
Quantitative Analysis - Rietveld
Phase 1 : "Moissanite 6H_27051-icsd" 33.2(3) %
Phase 2 : "Zirconium Boride (1/2)_30327-icsd" 64.3(3) %
Phase 3 : "Zirconium Oxide - T_85322-icsd" 2.53(7) %
Background
One on X 0(8000)
Chebychev polynomial, Coefficient 0 500(200)
1 -200(200)
2 50(130)
3 -60(70)
4 9(40)
5 -30(20)
6 8(11)
7 7(6)
8 18(3)
Instrument
Primary radius (mm) 300
Secondary radius (mm) 300
Simple axial model (mm) 20.04(9)
Corrections
Specimen displacement 0.025(2)
LP Factor 90
Absorption (1/cm) 150(40)
Miscellaneous
X Calculation Step 0.005
Finish X 109.98
Structure 1
Phase name Moissanite 6H_27051-icsd
R-Bragg 7.215
Spacegroup "P 63 m c"
Scale 0.00833(10)
Cell Mass 240.577
Cell Volume (?^3) 124.45(3)
Wt% - Rietveld 33.2(3)
Double-Voigt|Approach
Cry size Lorentzian 34(4)
k: 1 LVol-IB (nm) 22(3)
k: 0.89 LVol-FWHM (nm) 31(4)
Strain
Strain G 0.0(17)
e0 0.000(4)
Crystal Linear Absorption Coeff. (1/cm) 147.19(4)
Crystal Density (g/cm^3) 3.2100(9)
Preferred Orientation (Dir 1 : 0 0 6) 1.024(12)
PV_TCHZ peak type
U -0.22(15)
V -0.04(15)
W 0.04(3)
Z 0
X 0.10(7)
Y 0
Lattice parameters
a (?) 3.0819(3)
c (?) 15.130(3)
Site Np x y z Atom Occ Beq
C1 2 0.00000 0.00000 0.00000 C 1 5(2)
C2 2 0.33333 0.66667 0.16667 C 1 5(10)
C3 2 0.33333 0.66667 0.83333 C 1 6(11)
Si1 2 0.00000 0.00000 0.12500 Si+4 1 1.0(2)
Si2 2 0.33333 0.66667 0.29170 Si+4 1 1(10)
Si3 2 0.33333 0.66667 0.95830 Si+4 1 1(10)
Structure 2
Phase name Zirconium Boride (1/2)_30327-icsd
R-Bragg 2.169
Spacegroup "P 6/m m m"
Scale 0.1395(3)
Cell Mass 112.846
Cell Volume (?^3) 30.7242(9)
Wt% - Rietveld 64.3(3)
Double-Voigt|Approach
Cry size Lorentzian 490(40)
k: 1 LVol-IB (nm) 310(20)
k: 0.89 LVol-FWHM (nm) 430(30)
Strain
Strain G 0.1008(14)
e0 0.000220(3)
Crystal Linear Absorption Coeff. (1/cm) 690.53(2)
Crystal Density (g/cm^3) 6.09895(17)
Preferred Orientation (Dir 1 : 0 0 1) 0.9951(12)
PV_TCHZ peak type
U -0.0058(10)
V 0.0044(14)
W 0.0026(4)
Z 0
X 0.024(3)
Y 0
Lattice parameters
a (?) 3.16975(4)
c (?) 3.53102(5)
Site Np x y z Atom Occ Beq
Zr1 1 0.00000 0.00000 0.00000 Zr 1 1.15(2)
B1 2 0.33333 0.66667 0.50000 B 1 1.72(15)
Structure 3
Phase name Zirconium Oxide - T_85322-icsd
R-Bragg 5.039
Spacegroup "P 42/n m c Z"
Scale 0.00118(3)
Cell Mass 246.446
Cell Volume (?^3) 65.49(4)
Wt% - Rietveld 2.53(7)
Double-Voigt|Approach
Cry size Lorentzian 0(300000)
k: 1 LVol-IB (nm) 0(200000)
k: 0.89 LVol-FWHM (nm) 0(300000)
Strain
Strain G 0.31(4)
e0 0.00067(9)
Crystal Linear Absorption Coeff. (1/cm) 664.2(4)
Crystal Density (g/cm^3) 6.248(4)
Preferred Orientation (Dir 1 : 1 0 1) 0.457(5)
PV_TCHZ peak type
U 0.17(19)
V 0.10(13)
W -0.04(3)
Z 0
X 0.18(10)
Y 0
Lattice parameters
a (?) 3.5983(8)
c (?) 5.059(2)
Site Np x y z Atom Occ Beq
Zr1 2 0.75000 0.25000 0.75000 Zr+4 1 0.18
O1 4 0.25000 0.25000 0.46000 O-2 1 0.8
herisson
第4楼2021/05/24
谢谢老师的帮忙和回复。
其实是想请教一下这样“略有规律的”低角度峰强计算值和理论值的差距,会是哪些因素造成的呢?结构参数、或者峰形参数有误?
因为我所做的这一组4个样品里,精修的时候这个ZrB2相都有这个情况,所以比较困惑。怕是刚开始学习,基础知识不牢固,犯一些关键性错误。
再次谢谢老师的回复~
iangie
第5楼2021/05/25
如果测得峰强与模型拟合不符, 很大程度上是制样的问题
Rietveld 拟合模型是根据粉末衍射假设的. 你的样品晶粒应该足够细 才符合粉末衍射的假设. 就目前看来你的衍射峰峰宽很窄, 表明晶粒并不是很细. 如果可能的话把样品磨细再测, 所得强度可以跟符合模型.
此外, 把测量几何信息带入精修软件可以更好地拟合峰形. 包括你用的是Bragg-Brentano反射几何 还是毛细管透射? 是实验室XRD()测角仪半径,各种狭缝宽度等) 还是同步辐射?
herisson
第6楼2021/05/25
原来如此,学习了~
我的粉末样品确实不是很细,有40-50μm,下次再做的时候我磨细一点试试。
XRD是在实验室测得的,找老师要了仪器参数,不过没有仔细看测角仪半径、狭缝宽度之类,只关注了下波长和极化因子。
您说的几何信息是在添加仪器文件时import diffractometer constants选中吗?