

人来人往
第3楼2007/08/22
多谢 ken919 的关注。
下图为依次改变超声功率及时间所得的曲线:
1、超声功率为10,时间30s时加入样品,得红色曲线;d(0.5):17.116;; d(0.9):30.100
2、超声30s后测量; d(0.5):13.600 ; d(0.9):29.127
3、第2次超声30s后测量; d(0.5):12.153;; d(0.9):26.294
4、第3次超声30s后测量; d(0.5):11.982;; d(0.9):25.138
5、第4次超声30s后测量; d(0.5):11.115;; d(0.9):23.499
6、设超声功率为15,30s后测量;d(0.5):10.189;; d(0.9):21.818
7、第2次超声30s后测量; d(0.5): 9.401;; d(0.9):20.397
8、超声120s后测量; d(0.5): 7.675;; d(0.9):17.225
9、超声30s后测量; d(0.5): 7.355;; d(0.9):16.628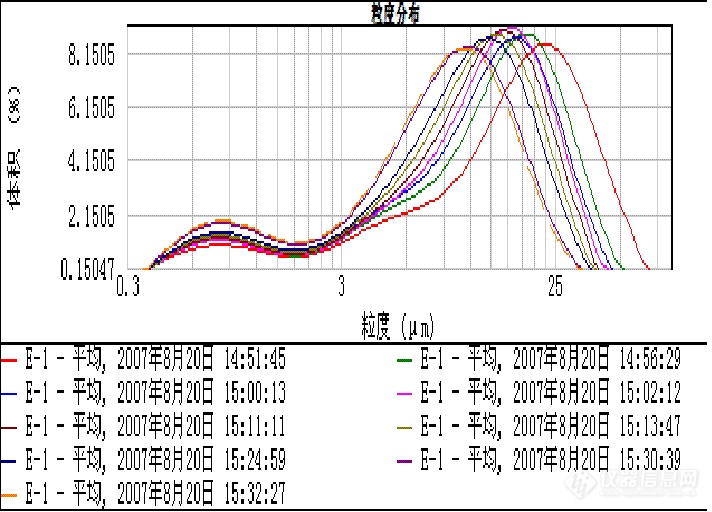
从测试结果看,很难确定那种条件时合适的。
另外,每次加超声后多进行几次测量的结果稳定,曲线可重合(没有上传),是不是说明该样品不需要超声就可以分散的很好?
对您的解答还有一些不明白的地方,望能进一步指点:
1、 残差不是用来衡量拟合度的吗?怎么是衡量你系统的洁净程度呢?
2、 如下图是多次清洗后,不加其他样品,单纯的水分散剂的的测试图形(遮光度0~0.2%),从不加超声到加2min超声到一直超声,分布图形杂乱无章。从这么多图形来看,水本身会不会对样品的测试结果产生的严重的影响?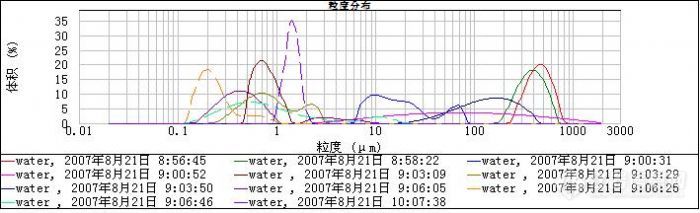
ken919
第4楼2007/08/22
不好意思,重量残差评估系统洁净的说法我也是听工程师说的/现在以前的贴子找到的说法是:残差是得出的粒度分布(即报告)按理论计算出的光强分布与实际检测器接受到的光强分布对比的差值,一定程度上可用来判断报告中光学参数的好坏,如果光学参数(折射率,吸收率)选的比较准确,则残差较小。值的大小由软件计算得出,与测试次数无关
上面的图是指一直超声后的测量读数还是重新加样的?要看超声后连续测量多次,每次间隔3分钟看看叠加图的变化 时间可以长一点
一般纯水分散剂是较洁净的,但会有一些微粒在里面但应作为背景可以扣除,所以对结果影响不大.
有时样品池有气泡也会这样,光路系统不干净也会这样,你主要分析背景就可以了吧,我记得ME一程师说过看原始数据图就是有背景和样品信号的那个图
人来人往
第5楼2007/08/22
谢谢ken919的回复。
第一个图形是同一个样品连续测试的结果。每次超声后会连续测试3x3次,耗时大约2~3min;然后再加超声再测试。每次超声后粒径会变小,超声停止后连续测试3x3次的曲线可重合。
水的那个测试图形:我觉得作为背景的话首先要求稳定,不然不能保证测试结果的一致性。可从上面的图形看,曲线是杂乱的,这会不会对测试结果造成干扰?
Malvern的工程师总说说我们用的水偏酸,对测试结果影响太大。我不明白微偏酸性的蒸馏水会对结果造成什么影响?容易产生气泡吗?
(我们测试的样品主要有硅微粉、氢氧化镁、氢氧化铝、滑石粉、有机填料等)

































