推荐厂家
暂无
暂无


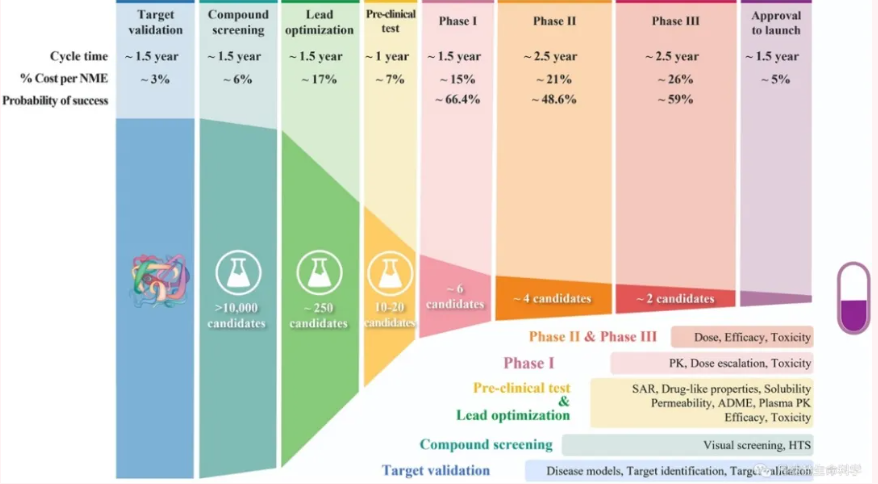



全部标题重视新药非临床安全性评价供试品的检测作者胡晓敏、冯毅、王庆利部门药理毒理学部正文内容 非临床安全性评价是新药开发中的重要内容,其耗时长(从一周到几年)、花费大(几十万到几百万元RMB),但其结果对于发现新药的毒性,预测临床安全性具有重要的意义。非临床安全性研究与评价贯穿于整个新药的研发过程中。供试品是非临床安全性评价获得可靠结果的基础,其质量和配制的准确性,直接影响非临床安全性试验结果及新药的后期开发。为了使新药安评的结果可靠,并避免在新药研发过程中出现不必要的失误和损失,在非临床安全性评价试验中应加强供试品检测。1 供试品检测的必要性 2005年国家局发布了多个非临床安全性试验技术指导原则,这些指导原则对规范我国药物安评试验,推进GLP的实施起到了重要作用。近来,不断有相关研究机构和专家反映,由于未能在有关的技术指导原则中要求开展安评试验的供试品的相应检测,致使安评试验结果具有不确定性。 目前我国创新药的研究与申请逐渐增加,为了使安评结果准确可靠,弥补非临床安全性试验技术指导原则中的对供试品检测未设置相关要求的缺憾,建议在安评试验中应加强供试品检测,避免出现不必要的失误和损失。2 国内外对供试品检测的要求 FDA、EMA在GLP规范中,对供试品的检测提出了要求。各大制药公司内部的SOP也对供试品检测有要求。 我国的GLP对供试品检测也有原则性要求。但由于在新药申报和相关技术评价指导原则中未对此有明确要求,故当前不是每个GLP试验室、或每个安全性试验都进行供试品检测。3 供试品检测的适用范围 安全性试验中供试品检测的要求,应该适用于所有新药研究。中药成分复杂,结构不清楚的成分多,但如中药一类(单一成分)可参考化药执行。欧美对生物制品也要求进行供试品检测,内容在化药的稳定性、均一性等的基础上增加蛋白含量分析和生物活性分析。对于生物制品供试品检测的要求,建议参照化药的方法,遵从Case by case的原则。4 供试品检测的内容 ①供试品的基本理化性质检验报告(包含来源、批号、纯度、浓度、处方组成(包括辅料)、稳定性、溶解性、有效期、保存条件等信息)。②若供试品需经溶解后(混合、混悬、溶解)给药,则应提供供试品在溶剂中的稳定性、均一性(非溶液体系)等检测报告(浓度范围需能覆盖全部毒理试验的浓度范围),以及配制后的供试品浓度分析报告。③针对检测供试品浓度和含量分析的方法学验证报告。5 供试品检测报告的提供 供试品检测方法的建立和验证,可以由申请人(含生产者)、GLP试验机构或第三方完成,或由其中的一方完成后转移至另外一方进行检测;由验证方提供供试品检测方法学验证的资料。 配制后的供试品浓度分析方法的方法学验证资料,应由完成配制后的供试品浓度分析检测的GLP实验室提供。 必要时对对照品进行分析,对照品的分析要求与供试品相同。如果对照品为上市产品,其基本理化性质等资料可以参照对照品的说明书和/或标签。
临床用药实例分析——执业药师继续教育资料
世界临床在研抗癌新药目录[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=156124]世界临床在研抗癌新药目录[/url]
 400-889-7796
400-889-7796
 留言咨询
留言咨询
 留言咨询
留言咨询
 400-860-5168转4645
留言咨询
400-860-5168转4645
留言咨询










 我要推广仪器
我要推广仪器
 下载APP
下载APP