推荐厂家
暂无
暂无
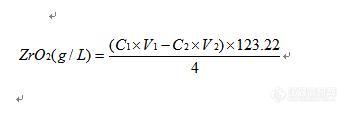
维权声明:本文为chenshaoj原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。现在硫酸锆溶液浓度分析方法大约有以下2种:1、 EDTA络合滴定法2、 重量法第一种方法是以二甲酚橙为指示剂,用EDTA标准溶液滴定至溶液由红色变为黄色即为终点,但通过长期运用,发现该方法指示剂指示终点颜色变化不明显,易照成较大误差;第二种方法是利用硫酸锆本身纯度很高,在配制的过程中又不用外加入任何化学物质即可溶解,所以取定量硫酸锆溶液,将其蒸发至干燥后,根据坩埚重量差即可计算出硫酸锆溶液的浓度,该方法较为准确,但大家都应该了解,重量法耗时很长,如果坩埚恒重不好的话,也容易造成较大误差;所以上述两种方法都具有一定的缺陷,不利于在生产中运用。所以我就改变思路,既然硫酸锆在配制的过程中并没有加入任何东西助溶,而硫酸锆又容易水解,其水解方程式如下:Zr(SO4)2+2H2O=2H2SO4+ZrO2 我们为何不能采用酸碱滴定其水解生成的硫酸从而计算出其氧化锆的浓度呢?所以就开始实验: 定量取一定体积的硫酸锆溶液(一般为2mL),加水稀释后以甲基红-亚甲基蓝为指示剂,用NaOH标准溶液滴定,溶液颜色由红色变为蓝色即为终点。 想法很好,但在实验的过程中发现了问题:由于在滴定的过程中,溶液的PH不断升高,所以硫酸锆也不断水解生成氧化锆,氧化锆会吸附指示剂,造成终点时变色不明显,而且颜色极易退掉,所以无法准确判断终点,换用其它酸碱指示剂(如酚酞等)也存在同样的问题。 既然溶液中的沉淀物会影响终点的判断,那为什么我们不把沉淀过滤后再来滴定呢?说干就干,通过实验发现,加入EDTA后硫酸锆也会水解(至今也没有想明白原理),而EDTA是中性化合物,不会对酸碱滴定产生误差,所以我就在硫酸锆溶液中加入EDTA溶液,等到硫酸锆全部水解后减压过滤,洗涤沉淀,滤液转移至三角瓶中以甲基红-亚甲基蓝为指示剂为指示剂,用NaOH标准溶液滴定,溶液颜色由红色变为蓝色即为终点;这次终点颜色很好判断,变色明显也不褪色,窃喜还以为成功了,但通过做几次平行样品后发现了问题:各次平行样品测定结果相差很大。这是什么原因呢?想来想去终于发现了问题:在加压过滤的过程中,溶液根本就没办法保证其全部将其水解生成的硫酸洗下来,而且EDTA是否会影响最后的滴定也不知道,另外这种方法也比较复杂,背离了我的初衷,所以该方法以失败告终。 后来又想了很多方法,比如直接用EDTA络合滴定时采用络黑T为指示剂等,最后发现也是存在指示终点不明显的老问题。 那我们可不可以采用反滴定法呢?那就用实验来说话吧:在硫酸锆溶液中定量加入过量的NaOH标准溶液,这时由于溶液呈碱性,硫酸锆早就全部水解了,而且溶液有一部分过量的NaOH,不过滤,以酚酞为指示剂,用盐酸标准溶液滴定至溶液红色刚好消失即为终点。通过实验,发现终点变色很明显,几个平行样品分析做出来结果基本一致,而且数据和定量配制的硫酸锆浓度刚好吻合。哈哈,实验成功,经过整理的方法如下:1、 原理硫酸锆水解后生成硫酸和氧化锆,先在硫酸锆溶液中定量的加入过量的氢氧化钠标准溶液,将水解出来的硫酸全部中和,过量的氢氧化钠以酚酞为指
[size=4] 在进行总氮实验时候,由于国标的要求:“当测定在接近检测限时,必须控制空白试验的吸光度A[sub]0[/sub]不超过0.03。”空白值偏高往往是总氮测定中不符合要求的一大难题。然而,影响总氮空白值偏高的原因,除了用水、器皿之外,过硫酸钾试剂就是最主要因素了。之前,我们也采购过不同几家试剂厂生产的过硫酸钾试剂,都做了试验,结果几乎都不尽人意,进口试剂也尝试使用过,但成本太高,没有选择。[/size][color=#DC143C]1.关于总氮试验中过硫酸钾空白值偏高,你们是怎么样选择的,能否说说你们的选择经历?2.你们购买哪家试剂厂生产的过硫酸钾,空白值可以达到多少?[/color]
硫酸高铈适合做cod快速测定仪的氧化剂吗?







 400-801-9298
400-801-9298
 留言咨询
留言咨询
 留言咨询
留言咨询
 400-860-5168转4207
留言咨询
400-860-5168转4207
留言咨询










 我要推广仪器
我要推广仪器
 下载APP
下载APP


