

hyban 2008/11/19
针对峰的漂移: 在更换流动相后,要尽量冲久一点,待系统充分饱和再进样; 尽量使用柱温箱,温度对出峰时间也会影响,尤其是昼夜温差较大的时候; 还可以考虑样品和对照穿插交替进样,因为试过遇到某些比较变态(只能这样形容)的检品,无论怎么折腾都会飘,无奈,只能样品和对照穿插交替进样,在报告书中附带说明。
风之彩 2008/11/20
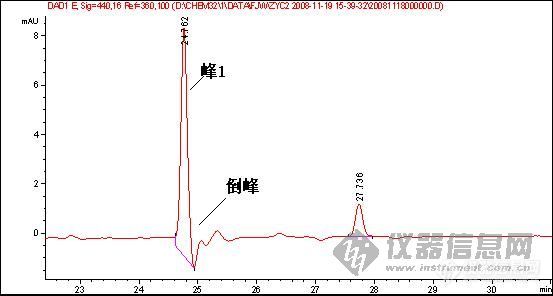
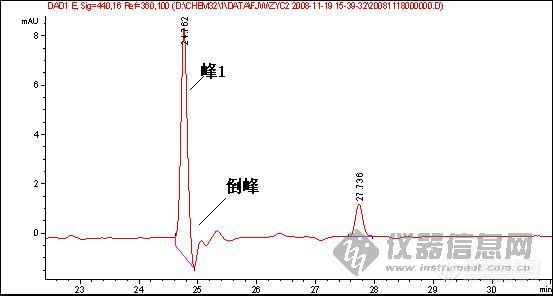
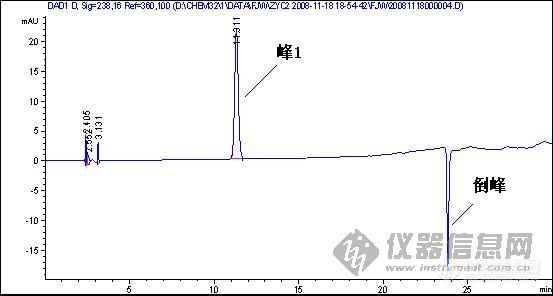
倒峰产生的原因: 1 信号线极性接反了或是参数设置成倒峰的; 2 走梯度走出来的; 3 溶剂峰 4 氘灯老化,流通管道或池不通畅; 5 样品和流动相中含有小颗粒,气泡等; [color=#DC143C]6 参比波长设置的不合适,如楼主的图,参比波长选择360nm,如果在出峰位置的某化合物在360nm左右的某一范围内有吸收,那么就可能出现倒峰[/color] 前面两张图的倒峰不是很明显,特别是第二个倒峰像是基线波动,样品峰后面的倒峰是不是样品溶剂的关系?换个溶剂看看?或是这个地方有干扰?调整保留时间看看? 第3张图可能都是第6种原因导致,建议把参比波长重新设置,或是干脆把参比波长关掉看看,可能就会好了。
tanghongmin 2008/11/19
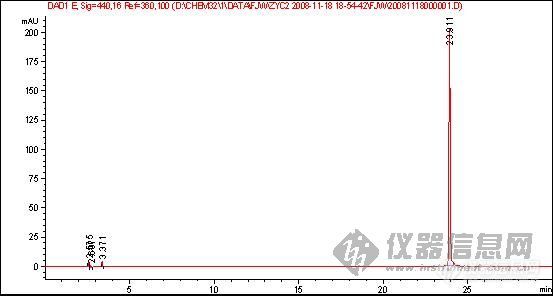
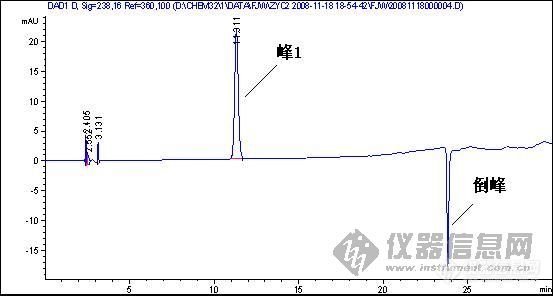
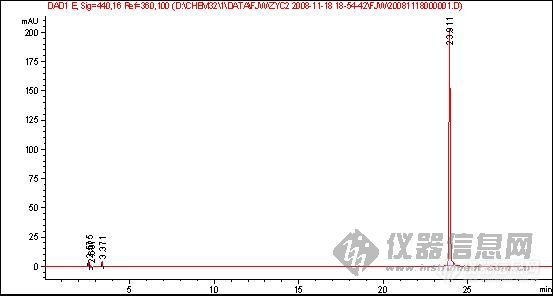
[quote]原文由 [B]03yx2[/B] 发表: [B][size=4][color=#DC143C][center]大家图谱来找茬(第三季)[/center][/color][/size][/B] [center]◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆[/center] [B] [color=#DC143C] 请您来分析: 1.产生倒峰的原因有哪些? 2.图谱中一个重要问题是倒峰,一个是离峰1很近的倒峰,一个是离峰1比较远,请问有什么区别?针对峰的漂移你有什么好的建议嘛? 3.如果要对图中的峰1进行含量测定,你觉得应该怎样改善条件才可以更好的进行定量分析.[/color][/B] [color=blue]〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓[/color] [color=#DC143C][B]图谱如下:[/B][/color] 1、样品峰1: [img]http://ng1.17img.cn/bbsfiles/images/2017/01/201701191651_626357_1632580_3.jpg[/img] ◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇ 2、该物质的对照品图谱: [img]http://ng1.17img.cn/bbsfiles/images/2017/10/20081119204739_01_1632580_3.jpg[/img] ◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇ 3、另一类倒峰: [img]http://ng1.17img.cn/bbsfiles/images/2017/10/20081119204834_01_1632580_3.jpg[/img] [size=2][color=#DC143C]【附录】凡参与活动者,必有重奖[/color][/size][/quote] 我基本赞成1、2楼的hyban的观点,但是对于已经25min出峰后的倒峰,应该不会是溶剂峰,溶剂峰出来比较早,一般在5min前,所以用流动相来配比定容供试液不会有作用 倒峰产生的原因我认为主要有两个,就如1、2楼的hyban说的检测器问题,光源不稳定,背景噪音大,如果我没有说错的话,你用的这个检测器是Agilent的紫外或是DAD(仪器用的比较多的板油做仪器耐用性的时候就能对比出来);另外一方面是当流动相极性比例发生大幅度改变时,泵后流动相由于死体积大或者泵来不及调整导致流动相混合不均匀,或者气泡没有完全除干净,温度未恒温等原因都会产生倒峰
ygx 2008/11/19
1.倒峰的出现,无怪乎有一下几个原因:试样的溶剂峰;试样中的杂质峰(且含量较大);流动相中有气泡;检测器性能不稳等。 2.第三幅图的倒峰应该不是溶剂峰。对于峰漂移的问题,对照品浓度和样品浓度应尽可能的接近,另外两者稀释的溶剂也应一致。 3.对于第三幅图,应该对峰1的定量不会带来多大影响的。 对于第一幅图,应该在流动相、检测器方面着手改进或完善:若是气泡问题,就对流动相彻底脱气;若是杂质问题,就改变流动相组成或配比,或者由恒流变梯度,或变换检测器波长等;若是溶剂的问题,就更换溶剂或用流动相稀释;若是检测器的性能问题,就降低检测器的灵敏度或更换光源等。
吹不动的浮云 2008/11/21
鉴于你的这个试验,在25分钟总会出一个倒峰,只是你的峰1的出峰时间在变,如果可以确定在25分钟是一个成分,我建议更换低吸收的流动相,定量需要重现,现在看来你做的样品不能够保证样品的保留时间重现,一是建议多稳定一段时间,二是,保证进样条件和进样量重现,三,保证样品不被污染和溶液成分的改变!(乙腈和水的吸收较小)
tanghongmin
第5楼2008/11/19
我基本赞成1、2楼的hyban的观点,但是对于已经25min出峰后的倒峰,应该不会是溶剂峰,溶剂峰出来比较早,一般在5min前,所以用流动相来配比定容供试液不会有作用
倒峰产生的原因我认为主要有两个,就如1、2楼的hyban说的检测器问题,光源不稳定,背景噪音大,如果我没有说错的话,你用的这个检测器是Agilent的紫外或是DAD(仪器用的比较多的板油做仪器耐用性的时候就能对比出来);另外一方面是当流动相极性比例发生大幅度改变时,泵后流动相由于死体积大或者泵来不及调整导致流动相混合不均匀,或者气泡没有完全除干净,温度未恒温等原因都会产生倒峰
迷失的精灵
第8楼2008/11/20
首先对你的标样和样品进行全波长扫描,在3D谱图里观察最大吸收波长
出现倒峰是因为你选的波长不合适,其物质吸收在流动相吸收之前造成的
可能原因有氘灯老化,流通管道或池不通畅,样品和流动相中含有小颗粒,气泡等。也有文献认为倒峰主要是水分引起。需要针对原因解决。
可以参考:http://www.sqyjs.com/Get/wenhua/0641117461583434.htm
一、泵的使用和维护
1、防止固体微粒进入泵内。因为尘埃或其它任何杂质微粒都会磨损柱塞杆、密封环、缸体和单向阀。泵的入口都应连接砂滤棒,输液棒的滤器经常清洗或更换。
2、流动相不应含有任何腐蚀性物质。例如氯仿、丙酮、二氯甲烷等,这些溶剂将会Peek树脂部件变脆;还有含有卤离子的流动相或能产生卤离子的流动相,如果使用了,分析结束后,应立即用去离子水清洗整个管路,因卤离子会腐蚀不锈钢部件或管路。流动相含有缓冲盐不应停泵保留在泵内时间过长,由于蒸发甚至由于溶液的净置就可能析出晶体。
3、泵工作时要注意流动相不要被用完,否则空泵运转会加快磨损柱塞杆和密封圈。输液泵的压力不要太高,否则会使密封圈变形,降低密封圈使用寿命。
4、在使用双泵过程中,如果不是梯度洗脱,尽量先混合。如果一个是有机溶剂,一个是含盐溶液,在用双泵的时候,有可能会析出晶体。
5、在使用缓冲盐作流动相实验结束之前和结束后,都要用含10%甲醇水溶液冲洗管路,不能用纯水冲洗,因纯水会对柱子造成损坏。进样阀要用适宜的溶剂进行清洗。
二、色谱柱的使用和维护
1、避免压力和温度的急剧变化以及机械震动。在进样的时候,进样阀扳手不能转动太慢,更不能停留在中间,如停留在中间,泵内压力剧增,这时再转动到进样位置时,过高的压力会损坏色谱柱。如出现机械震动(掉在地上)则会改变填料的充填状态。
2、在使用色谱柱过程中,不应直接从有机溶剂到水,反之,也不要从水直接改变到有机溶剂。
3、色谱柱一般不能反冲,除非生产者指明。
4、冲洗色谱柱时,每种溶剂至少冲洗50ml。
5、装在机子上的色谱柱,应每隔4~5天冲洗15分钟,可以避免长菌,保护色谱柱。
6、PH的调节尽量在2~9之间
7、保存色谱柱时,柱接头要拧紧。
三、输液泵产生 故障及排除方法
1、没有流动相流出,又无压力显示。原因可能是泵内有大量气体,这时打开排液阀,使泵在较大流量下运转,将气体排出,也可用一个针筒在泵出口处帮助抽出气体。另一个原因使密封圈损坏。
2、压力和流量不稳。原因①可能有气泡;②单向阀污染,可卸下单向阀,浸入异丙醇内超声清洗;③沙滤棒内有杂质或微生物堵塞,可将沙滤棒浸入流动相中超声清洗,也可将滤头放入4mol/l硝酸溶液中,迅速除去微生物。也可能是盐堵塞,可放入水中清洗;④在线过滤器堵塞⑤进样阀损坏;⑥密封圈性能不好。
3、压力过高。①管路堵塞;②保护柱堵塞;③检测池堵塞,发生情况立即停泵,否则会损坏流通池。
四、与检测器有关的故障及其排除
1、流通池内有气泡。如果有气泡连续不断通过检测池,将其噪音增大。如有较大气泡,则会在基线上出现许多线状"峰",可加大流量排出气体。如果检测池有停留的气泡,可采用突然增大流量的方法排除气体(不连接色谱柱)。
2、检测池被污染。检测池被污染了,可能产生噪音,或基线漂移,可以使用适当溶剂清洗,如果污染严重,就可以用1mol/l硝酸溶液、水和新鲜溶剂进行清洗。
3、光源灯出现故障。检测器的光远灯不能满足正常样品分析时,可能产生严重噪音污染,基线漂移,出现异常峰,甚至基线不回零。
4、出现倒峰。如果流动相有紫外吸收的杂质,使用紫外检测器时,就会产生倒峰,必须用高纯度的溶剂作为流动相。检测器的极性接反了,也会出现倒峰

































