三聚氰胺质控考核小记
一、前言
最近完成了一次质控样品的考核,项目是三聚氰胺。之前一次做三聚氰胺质控,还是在2008年三聚氰胺事件之后的应急质控,据同事回忆,当时的样品就是从市场上采集的问题奶粉,在发盲样时也提供了参考检测方法,该方法应该就是后来的GB/T22388-2008。同事用的是液相检测法,前处理采用瓦里安PCX固相萃取小柱净化,方法回收比较稳定,但达不到80%,基本上在70%左右。因此,在报结果时,同事比较纠结是否折算回收率,最终的结果还是把回收率考虑在内,考核也通过了。不过,当时质控样中的三聚氰胺的浓度较高,大概是目前常见三聚氰胺质控浓度的十多倍。同时,同事说那次三聚氰胺样品出峰前,存在一个较小的干扰峰,只不过是因为三聚氰胺浓度较高,因此小的干扰峰对结果影响不是很大。
二、液相方法
后来,我利用国标方法,发现有的加标样品确实存在干扰峰的情况,特别是浓度不高的时候,干扰峰对结果还是有一定的影响。于是将流动相比例做调整,有机相比例适当调低,使得三聚氰胺峰能够尽可能与杂质峰区分开来,于是得到的下面的图谱,但峰形较对称,但缺点是分析时间较长,达到25min以上。

三、液质方法
目前来看,三聚氰胺的方法比较成熟,利用国标或者在国标的基础上,对前处理做适当修改,例如蛋白沉淀的试剂以及固相萃取小柱的选择。22388中的第二法是液质法,我也利用液质联用仪开发了乳制品中三聚氰胺的检测。一开始用的是外标法,在优化方法时采用的是同一种基质的空白奶粉,因此,发现三聚氰胺存在较强的基质效应,国标中液质法测定三聚氰胺用的是基质加标曲线,考虑到了基质抑制对检测结果的影响。同时,也注意到不同基质乳制品所引起的基质效应的差别,这时就存在基质加标曲线的代表性问题。放大了说,这也是大部分液质外标法,在定量时所不能回避的问题。
因此,在国标的基础之上,我购买了三聚氰胺的13C标记的同位素内标,采用内标法定量。采用的色谱柱是HILIC 1.8um,液相部分是UHPLC,分析时间很短,只有1.5min,相对液相方法来说,UHPLC-MS/MS方法大大缩短了分析时间,能够较快的得到实验结果,并很快调整实验方法,同时也大大提高了分析灵敏度。但基质效应仍然存在,对检测结果影响也较大,因此内标法能够很好的去除样品前处理和基质效应带来的影响。
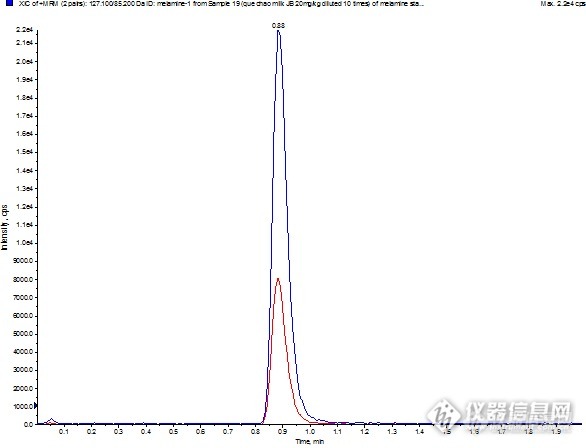
四、曲线范围
这次质控样共两个样品,经初步检测,两样品浓度有差别,但差别较小。配制了0.1~10.0mg/L的纯标标曲,用国标中的前处理方法,分析得到初步结果:1.0mg/kg附近,小数点后第二位不能确定。
初步确定了浓度后,可以调整标准曲线浓度范围,国标22388-2008中三聚氰胺基质加标曲线是0.01、0.05、0.1、0.2、0.5mg/kg,但稀释效应可能会带来一定的影响。我采用0.1、0.2、0.5、1.0、2.0、5.0、10.0mg/L(内标1.0mg/L)的纯标标准曲线。曲线中内标浓度是曲线中间点附近的浓度,这样能使得内标校正的误差减小。同时,样品中的浓度最好也处于标准曲线的中间部分。
结果发现,加标曲线和纯标曲线的线性都很好,两者的回归曲线相当,计算结果差别在小数点后第二位,大概0.02的样子,所以带内标的纯标曲线应该能够替代加标工作曲线。


五、基质效应、蛋白沉淀
第二次实验过程中,遇到了第一次实验没有发现的问题,即质控样中的一个样品出现蛋白沉淀效果不好的现象,随之而来的是过滤纸速度下降、过MCX的SPE小柱时有严重的堵柱情况,使得实验室时间拉长,对结果可能会有一定的影响。此外,这次实验还发现两个质控样的基质效应相差大约5倍,也就是有一个样品的基质效应特别强。因此,在这种情况下,如果采用外标法做,在配制基质加标曲线时,该曲线是否能够代表两种基质的质控样品?定量准确性又如何把握?是否要使用向质控样中加入标准的方法,确认质控样的回收率?实验中有一个样品的测定结果发生了变化,变低了,具体原因不详,可能与蛋白沉淀效果不好有关。
第三次实验,这一次沉淀效果较好,使用的试剂与国标一致,三氯乙酸的浓度和量都没有改变,也有人提出用高一点浓度的三氯乙酸,或者用乙酸铅等其他沉淀剂沉淀蛋白,但由于时间问题,没有一一尝试。这次实验过程中有一个巧合,上午开始处理样品,做到三氯乙酸、乙腈加完、超声、振荡提取这一步后,正好到午饭的时间,于是就放在那边,大约半个小时左右,回来后再离心,发现沉淀效果较好。难道加完沉淀剂后,还要让其反应一段时间?是由于冬天室温较低、所以蛋白沉淀的反应时间较长?
六、空白干扰
实验中还有另外一个头疼的问题,就是空白干扰。在进纯溶剂的时候,没有发现空白干扰峰的情况,但只要经前处理后,空白溶剂、空白奶粉经处理后都会出现空白干扰。在计算加标回收率时发现,如果不扣除空白,回收率会会比100%高出一些,扣除空白后,基本上在100%左右。实验中采用的是塑料离心管,也有同仁提出要采用玻璃试管,我没有采用玻璃试管的原因主要是怕离心时容易破裂。同仁也提出,就算用塑料离心管的话,也不要重复利用,怕有残留。这一点我倒是做到了,但结果还是会出现干扰峰。无奈,最后只能一一扣除空白。纯标曲线不需要扣除空白,加标曲线扣除空白样品中的空白,质控样扣除空白溶剂经处理后的空白。注:空白中都加入了同等浓度的内标。

 之前也看到有版友提出是固相萃取小柱的问题,我用的是OASIS的MCX小柱,其他的小柱还没进一步尝试,欢迎版友提出宝贵经验。
之前也看到有版友提出是固相萃取小柱的问题,我用的是OASIS的MCX小柱,其他的小柱还没进一步尝试,欢迎版友提出宝贵经验。
七、实验条件
1、液相:液相采用的是国标的流动相,等度洗脱,HILIC 1.8um的色谱柱,实验发现,等度洗脱保留时间、响应均较稳定,但HILIC柱使用前,需要给足时间平衡色谱柱。此外,与一般的色谱柱一样,在分析完之后,要用同等比例的乙腈水冲洗色谱柱,并保存在90%有机相中。

2、质谱:采用ESI正离子的MRM扫描,MRM离子对、离子源参数以及质谱图谱详见下方图谱:





总的来说,这次质控主要遇到的问题还是空白干扰、基质效应、蛋白沉淀效果的问题。后两者通过内标定量、前处理步骤的微调,得到了解决。但空白干扰问题依旧存在。我同样做液相时,可能是由于液相检出限的问题,没有出现样品空白干扰的问题,但液相会存在样品杂质峰与目标峰难以分开的问题,容易出现假阳性,并且为了得到更好的分离,我把分析时间拉得太长(25分钟),因此这次的最终结果没有采用液相定量,而是同位素稀释的液质联用法。
欢迎各位版友提出宝贵建议,此外,也顺便说下,这次样品的浓度对于液相而言,确实不高,对于液质而言,由于基质效应的不同,还是尽量采用内标法。



































