仪器信息网APP
选仪器、听讲座、看资讯
立即体验
APP内打开
回版面
2
4
4
拍砖
举报
取消
发布
当前位置:
仪器社区
>
色谱
>
液相色谱(LC)
>
帖子详情
庐山面目真像大白——0701
一片枫叶
2016/07/07
私聊
液相色谱(LC)
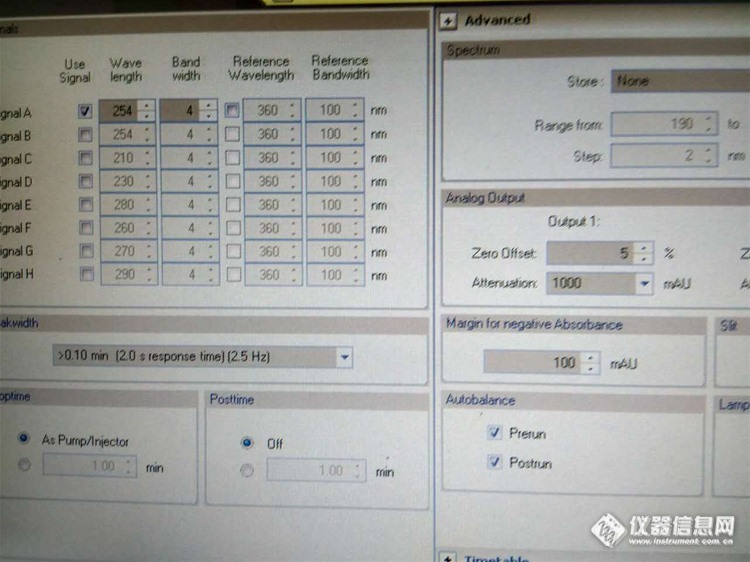
1:数据完整性如何做的——管理权限
答:1:每台电脑都要设置登录权限,实验软件要开审计追踪,没有审计追踪的,每次图谱的处理积分方式什么的都要在仪器使用记录上写清楚。
2:软件审计追踪,要设置用户.密码。以示只有你可以登录到该软件进行实验或者数据处理。
3:比如不能改时间,你做个改时间的操作,会提示要求管理员权限,改时区就提示不允许,然后你把这过程截屏
4:电子数据,每半年归档保存一次,存放在移动硬盘里,然后交给QA,做好电子数据归档的记录。
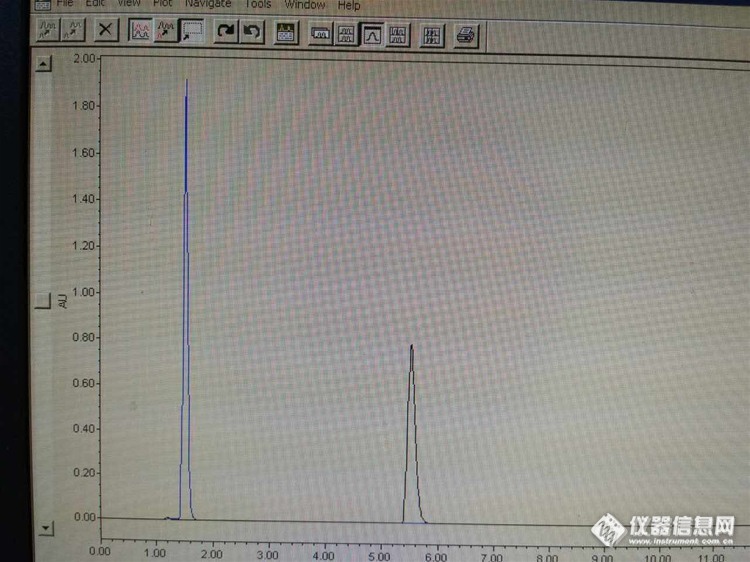
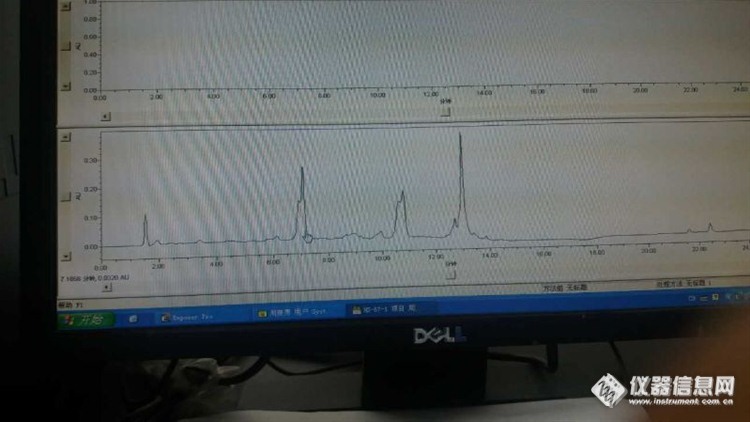
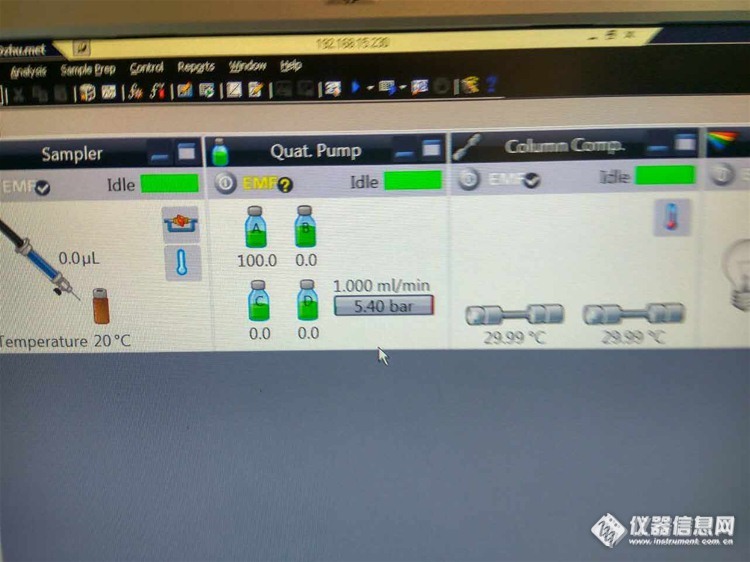
2:峰比正常情况下提前是 什么原因——蓝色的是反常的,黑色的是正常的
我把两个通道露出空气外面,以2ml/min的流速purge两通道, 发现a通道比B通道气泡走的快一倍了。已经排除了操作、方法设置等原因…
答:结论:峰提前的问题已经解决,是其中一个通道滤头损坏导致吸液较慢,从而使整个序列的流动相比例发生改变,致使峰提前。另外,网友有提到,比例阀故障也会引起此现象。检查故障时,确认比例阀无误,更换新的滤头,图谱恢复正常。
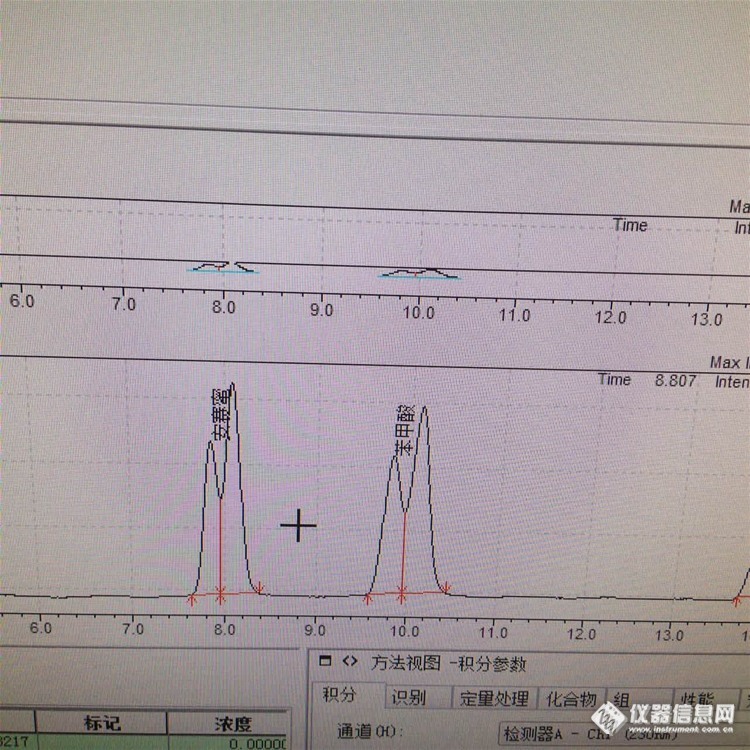
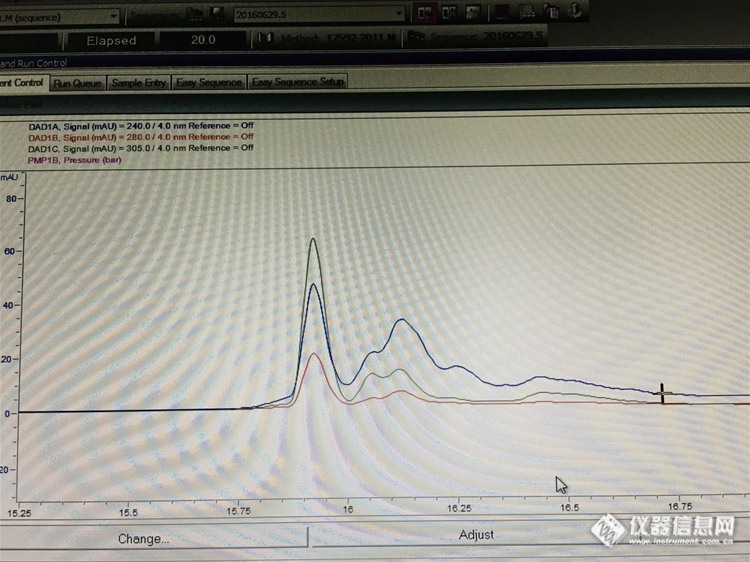
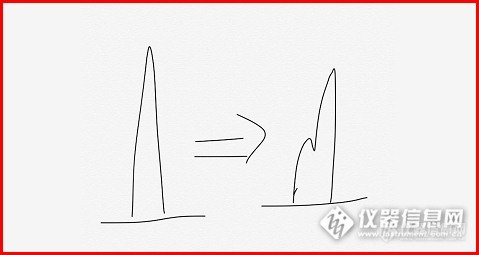
3:峰分叉的原因——甲醇➕乙酸铵(5%+95%),
答:推测:1:会不会一些成分没有洗脱下来,一直进样导致柱效降低——新色谱柱
2:溶剂选择不对——之前走样品正常
3:样品没有过膜或进样过载——样品已过滤减少进样量没有效果。
4:柱子接头不好有死体积——峰型不好或分叉
5:可能是由于样品在流动相中的溶解性差,样品在色谱柱头沉积致使柱头污染,造成柱压升高和峰分叉。
结论:刚才把保护柱芯拆掉了,没有分叉——保护柱污染
为了急于分享,系统没有走稳就进样了,图中出现了基线不稳和压力波动的现象,但分叉的问题解决了。
4:仪器:watersUPLC-ELSD-TUV。检测物:黄酮类化合物。
这三个图分别为进样3uL,4ul,5ul的图,是不是峰与周围的没分开,第一和第二的大峰随进样量加大出现这种情况会有可能是什么原因?
答:推测:1:只从图谱看是进样量的问题了。
2:前一针遗留杂质也不定, 你得连续进三针,然后间隔若干分钟后再进样,才能确定是不是杂质了。
结论:进样量过大——又进了个2微升的感觉峰形还可以。
5:我的样品浓度是1mg/ml进10ul,主峰是900,我稀释1000倍,那么现在相当于0.1%的水平的主峰,S/N=10,这个水平的定量限,是不是高了一点,常规应该是0.05%吧?
答:实验:1:稀释1000倍,即进样浓度0.1%水平的s/n=13
2:推测:稀释到0.05%水平的话,s/n=6.6。如果进样15ul的话,s/n应该能达标到10。但是按15的进样量的话,100%水平的浓度,主峰会大概到1400,问题就在这里,超过1000。waters的峰纯度鉴别不超过1000才准,所以有点冲突。
结论:要根据组份含量不同。安全指标,有限量和检出限。营养指标要测定准确,在标曲范围内
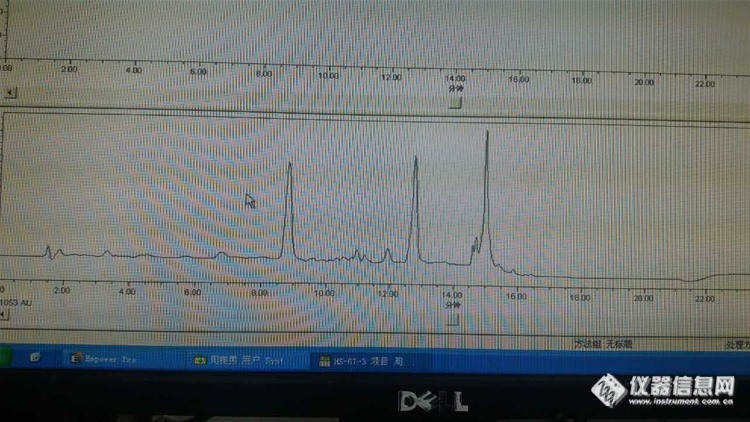
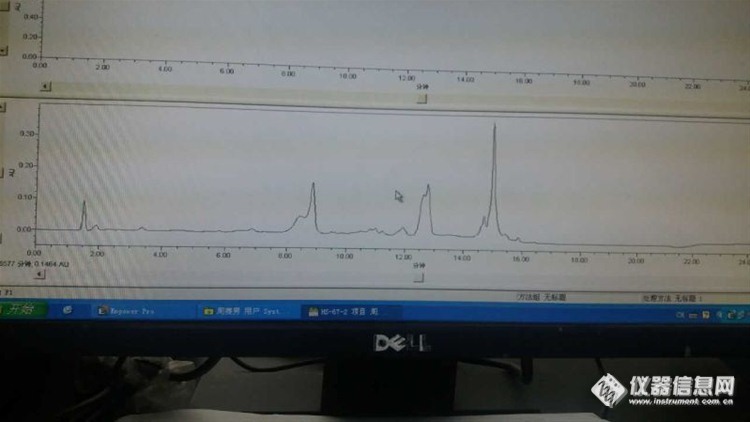
6:单标走出来这个样子,什么情况?
样品基质复杂,待测物出峰处有很多干扰物质,没法定量,走纯溶剂,什么都没有,以前走的很干净的,现在单标不应该走成这样。
答:推测:1:储备液储存太长发生了变化——杂质都是那么一致,所以我觉得不是仪器的原因,而且峰型也很好,这种情况下,我认为是标样原因。
2:还有就是改变样品的处理方法,或者使用质谱——待测物在杂质里面,根本找不出来,不像
气质
可以提取离子或用SIM模式。
结论:问题还没有彻底解决,我用原来的方法进了一针纯甲醇,发现正好在我出峰的位置有个杂质峰,我以为是溶剂污染了,重新打开一瓶色谱级甲醇,问题依旧。然后把方法改了,目标峰和杂质峰分开了,但是峰形不太好,有些拖尾。然后进样品,样品杂质很多,目标峰依然混在杂质中,无法定量。不知道进纯溶剂为什么会有杂质峰,以前是没有的。难道柱子被污染了?最后我用
气质
定量的。
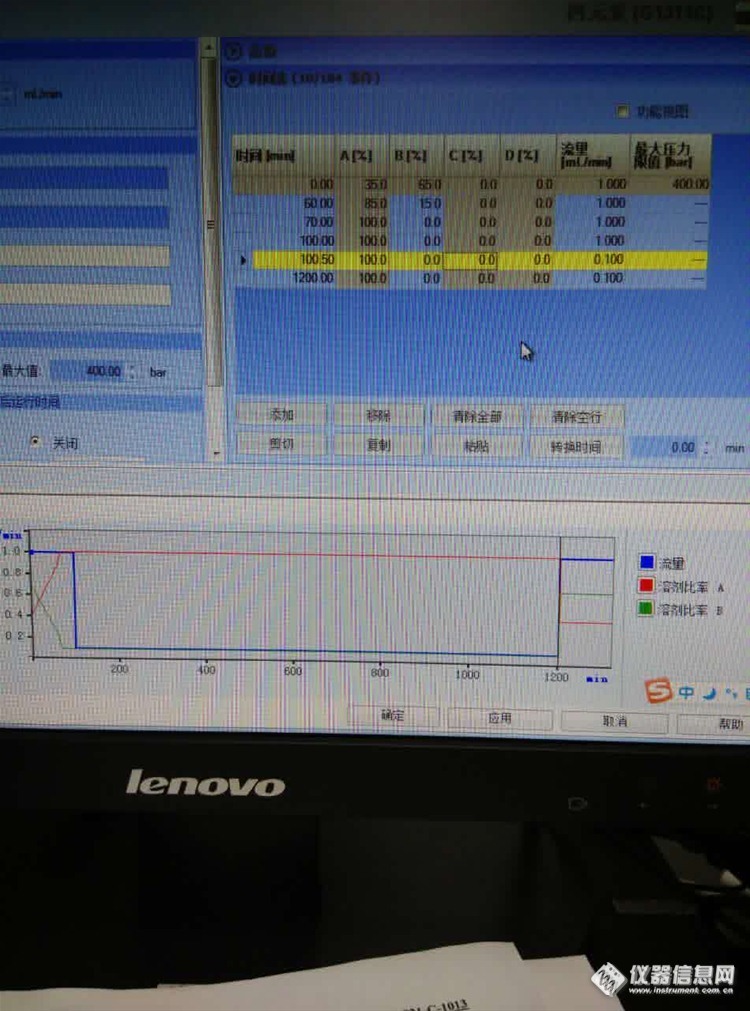
7:请问用完缓冲盐之后清洗程序怎么设置,为什么我设置梯度结果系统不按梯度走,安捷伦1260手动进样器,打到充样位置,纳闷呢,今天早上过来看,就是按梯度的第一个比例和流速在运行。
答:1:方法命名有没有仪器不识别的字符,方法运行结束后仪器是standby 还是shut down——当时是直接在仪器设置里面设置了方法,然后另存为,然后就直接搬动进样器让他运行设置的程序。要确保冲洗方法正常运行,还有一个shut down的程序,这个要设置正确。
2: 第一个梯度时间60分钟,你想啊,样品采集时间是有限的吧,梯度时间超过了样品采集时间,以后的时间怎么会执行梯度设置呢——采集时间应等于梯度最终时间。
3:没有单独调用设置里的这个方法。第一个程序是你的样品方法程序吗,进最后一针的时候有没有确保冲洗方法正常开始运行呢——以后一定要确保冲洗方法正常运行了才离开,还有一个shut down的程序,这个要设置正确。
8:安捷伦
液相
,灯能量合格,基线波动还是很大,流动相:正己烷,压力稳定,没有接柱子。
推测:1:流动相:正己烷,刚换水,没有接柱子,压力低——由正相色谱流动相向反相色谱流动相必须有中间过渡溶剂(比如:异丙醇),没有接柱子自然压力低。
2:灯设置是开着的,流通池内有气泡。
结论:流通池的问题。
9:出现前肩峰——不是柱子,同样的样品,第一针会 第二针不会,第三针可能又会,第四针第五针又好了。
说明:因为前几天调针位置时候出现高度没调好,微漏,歇了一晚上 ,第二天重新调,调整好了,不漏了,但是我色谱图出现了这样的问题,后来连续进了十针,看看精密度,结果前四针出现类似情况,第五针之后就好了.rsd很差,接的质谱,sim模式的峰面积,偏差太大了 有几倍的关系。
推测:1:怀疑依然是针位置没调好,针也有一点点弯了感觉。
2:针座密封圈不密封 对色谱图会产生影响——漏液样品走丢,对峰响应有影响。
结论:峰分叉的问题:工程师建议把autosample 里needle stroke 进样针冲程由52改成54,后来继续走就没问题了,仪器精密度还没跑,还不确定是不是改之后会解决这个问题,还没做对比。
10:最近哪台uplc每天早上开机后会自己断电重启,量了电源电压,稳定时是24v,断电重启电压就低很多,是不是供电电源坏了,有没有遇到过的
推测:1:是主机重启还是检测器重启——高压电源问题。
11:
液相色谱
中洗针的工作原理
答: 1: 自动进样器型号不一样,方式不同——岛津和安捷伦的自动进样器是有个小瓶,针到瓶子里沾一下,清洗外壁,赛默飞的自动进样器有单独的外壁清洗液,有个蠕动泵 。
液相
R通道洗,也有可以流动相洗的设置, 赛默飞的是D通道。
2:洗针功能可以把样品残留降低到最低限度,这里的洗针—仅仅是洗针的外侧,即将其在某瓶溶剂中蘸一下而已,针的内壁不需要清洗,应在瓶中盛放能溶解样品的溶剂。针内也有清洗的,洗针时选择inject模式。
3:安捷伦高性能自动进样器——有一个带有蠕动泵清洗孔能够清洗针的外壁,对非常敏感的样品分析来说,这可以减少已经很小的交叉污染,应用洗针的溶剂瓶可放在溶剂柜上,洗针产生的废液通过一个废液管排放。
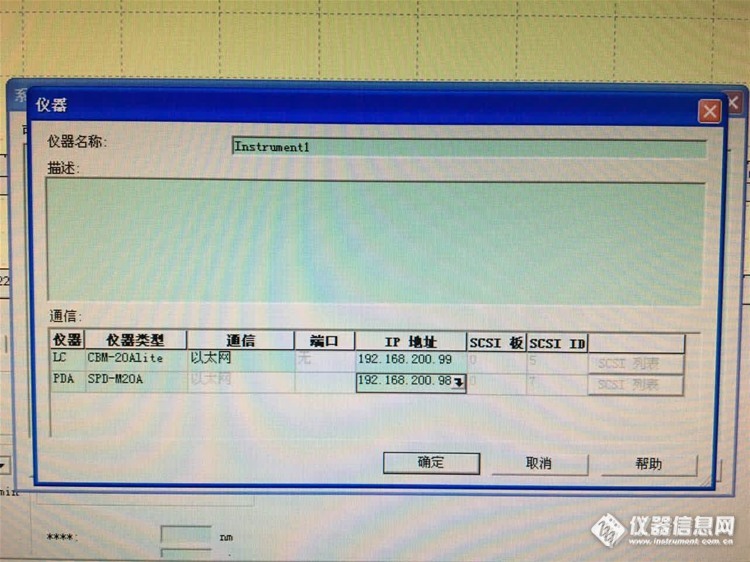
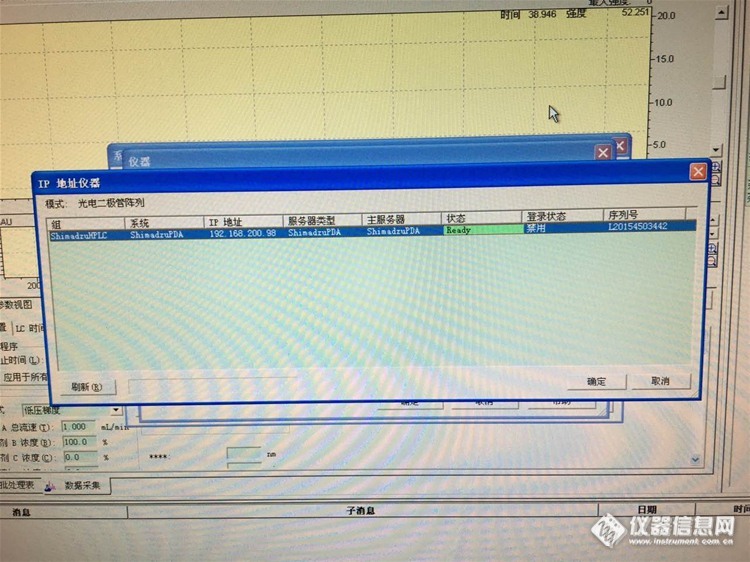
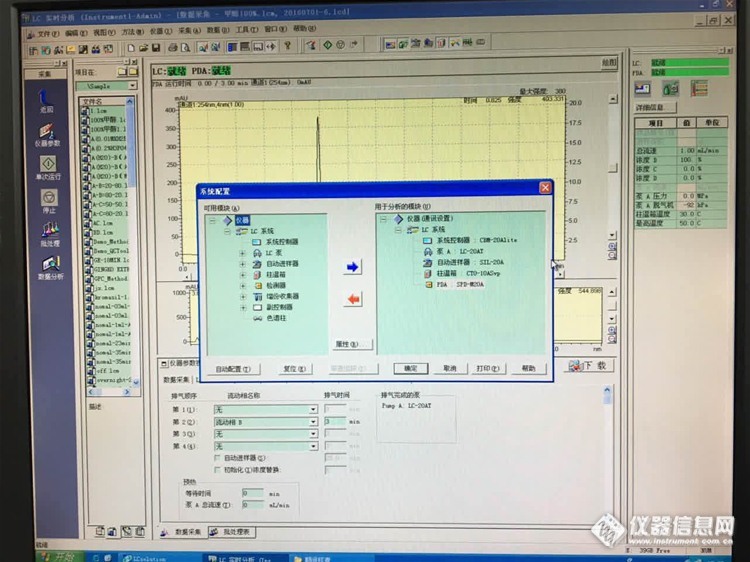
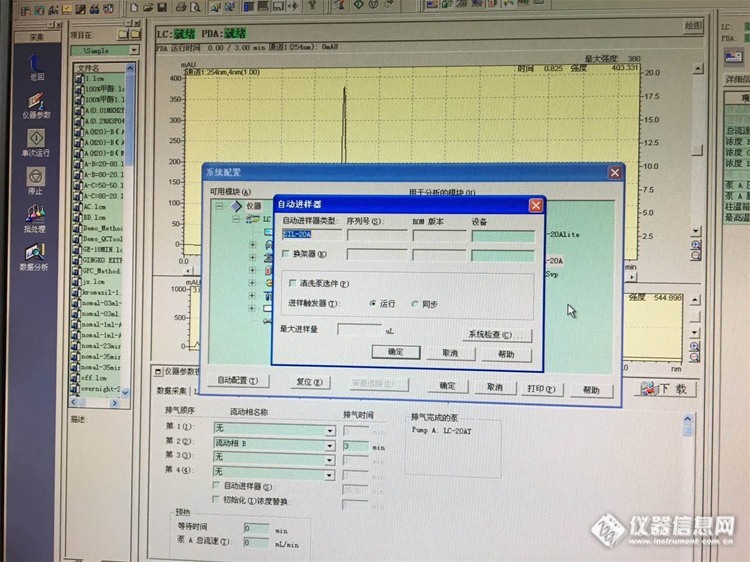
12:岛津LC-20A检测器被禁用,怎么办?
推测:1:查看信号线——网线接头没有松动,而且可以走基线,现在也不能控制自动进样器进样了。
2:没配置进去咋控制自动进样器——有基线并可以绘图。
3:模块故障——基线是检测器信号吧,自动进样器是独立模块,也有独立的信号,你尝试一下能否控制洗针的清洗动作。
4:如果可以控制清洗和调流量,唯独不能进样,再看看是否是卡槽复位问题。
5:更改了电脑IP,PDA就启用了,但是未连接状态。
结论:仪器神奇般的活过来了!方法是更改一下仪器配置中的自动进样器的状态,同步改运行,如下图……
至于PDA的禁用状态,对仪器使用没有影响~依旧如下图
相关话题
1
HPLC检测BPA
2
三聚氰胺用什么方法测比较好
3
【液相色谱之家】老师们好,请问酸性红和酸性红73是一种物质么……
4
【液相色谱之家】 请问方法检出限和定量限怎么做啊
5
液相梯度出的尖峰
+关注
私聊
一片枫叶
第1楼
2016/07/17
应助达人
顶一下。
0
发表回复
+关注
私聊
妮妮118
第2楼
2017/08/22
非常好的帖子
0
发表回复
近期热榜
赛默飞实验室产品焕新计划有奖调研!
【七月征文】不一YOUNG实验“猿”
报名开启:ICS2024第十三届光谱网络会议!
推荐收藏!盘点中药材及饮片检测解决方案
热门活动
甄选国优仪器,助推设备更新
采购咨询618活动:1000元奖励等你拿
猜你喜欢
最新推荐
热门推荐
更多推荐
SSI的说明书
悬赏帖
2013/04/19
示差检测器
已应助
2022/10/06
【转帖】使用waters的AQC衍生氨基酸的人注意了
2010/04/09
[共享]:戴安的资料
2006/04/03
【论坛劳模】 自动进样器低压阀泄漏的维修
原创
2014/05/05
在不换柱子的条件下,如何分开果糖和木糖
求助
2019/07/19
主动阀,拔下来还是拧下来?
第九届原创
2016/09/19
waters e2695 sealwsh管,插泵头的地方漏液,怎么办
求助
2017/08/03
【我们不一YOUNG】+气相色谱出现基线噪声大的原因
分享
2024/07/08
【我们不一YOUNG】+液相色谱使用过程中出现基线飘逸的原因及处置方式
分享
2024/07/08
【我们不一YOUNG】+液相色谱泵压不稳的原因
分享
2024/07/08
【我们不一YOUNG】+液相色谱泵中系统压力升高的原因及处置办法
原创
2024/07/08
【我们不一YOUNG】+液相色谱泵不送液的原因有哪些?
原创
2024/07/08
【我们不一YOUNG】液相色谱压力异常怎么办?
原创
2024/07/07
【我们不一YOUNG】高效液相色谱仪
原创
2024/07/07
【我们不一YOUNG】液相如何改善峰形与提升分离度
原创
2024/07/06
检测报告一定要有判定结果吗?
求助
2024/07/01
气相FID重现性不好原因
求助
2024/07/01
求助安捷伦GC7890B,EPC无响应解决办法
求助
2024/07/01
‘有奖问答’对错题’:用抽样方案(30,0)对产品批进行连续验收,当批不合格品率为1%时,方案的接收概率为73.97%,则平均检出质量为( )。
讨论
2024/07/05
HJ1332便携非甲实际应用问题
讨论
2024/07/01
如何做好体系工作?
分享
2024/07/01
多人主检的报告,检测审核应该怎么签字
求助
2024/07/05
EI源的灯丝镉厂家通用吗?
求助
2024/07/03
红枣香精中的未知物求助
悬赏帖
2016/07/07
未知物定性求助
悬赏帖
2016/07/07
UPLC 求助 请问这是什么问题
求助
2016/07/07
邻苯二甲酸二(2-乙基)己酯标准品质谱图无法与谱库匹配
求助
2016/07/07
凯氏氮比总氮高
讨论
2016/07/07
HPLC分离度的计算及其影响因素
分享
2016/07/07
中药制剂质量标准的制定
求助
2016/07/07
怎么排除ICP-OES测试金属中铅含量的干扰
咨询
2016/07/07
品牌合作伙伴
岛津
日立科学仪器
珀金埃尔默仪器(上海)有限公司(PerkinElmer)
日本电子株式会社
丹纳赫
安捷伦
赛默飞世尔科技
普析通用
欧波同
天美
天瑞仪器
德国耶拿
海能技术
马尔文帕纳科
磐诺科技
上海仪电科仪
梅特勒托利多
聚光科技
莱伯泰科
盛瀚
多宁生物
丹东百特
科哲
卓立汉光
屹尧科技
华谱科仪
宝德仪器
优莱博
HORIBA
布鲁克核磁
举报帖子
执行举报
点赞用户
好友列表
加载中...
正在为您切换请稍后...