摘要: 目的 评价不同一级分析方法对基于近红外光谱的透明质酸分子量模型影响。 方法 首先采用特性黏度法和多角度激光散射结合凝胶渗透色谱法测定透明质酸粉末分子量,作为一级数据,并同时采集样品的近红外光谱;采用K-S方法对样品集进行划分,对重均分子量和黏均分子量模型进行评价,同时考察非线性算法SVM和线性算法PLS对建模的影响。 结果 黏均分子量模型优于重均分子量,PLS模型均优于SVM模型。结论 不同的统计学分子量对建立模型有影响,同时分子量与近红外光谱之间的线性关系多于非线性关系。
关键词:近红外光谱分析技术 透明质酸 重均分子量 黏均分子量
透明质酸(hyaluronic acid,HA)是由重复双糖单位连接组成的链状聚合物,是细胞外基质的重要成分。由于其独特的黏弹性和生理功能,已被广泛应用于医药、美容、医用功能性材料和保健食品等方面。分子量是表征透明质酸的重要参数之一,不同分子量的透明质酸具有不同的理化性质,决定了其具有不同的用途。目前分子量测定的方法有特性黏度法 色谱法 电泳法 沉降平衡法。透明质酸是一种聚合物,具有多分散性,分子量表征均为平均分子量。采用的平均方法不同,所得的平均分子量就不同。如重均分子量是分子重量统计平均的分子量,黏均分子量是以黏度推算得到的分子量。本实验以透明质酸为研究对象,采用近红外光谱分析技术结合化学计量学算法,以黏均分子量和重均分子量分别建立透明质酸分子量模型,对模型进行评价,同时考察分子量与近红外光谱间的线性关系。
1材料
1.1 试剂
0.2mol/L氯化钠溶液(称取氯化钠11.688g,加水溶解至1000ml。),
1.2 仪器
Luminar 5030型便携手持式近红外光谱仪(美国Brimrose公司),InGaAs检测器,SNAP!光谱采集软件、The Unscrambler化学计量学分析软件(CAMO公司)、matlab2009化学计量学分析软件,自制支架及样品池。SYD-265D-1石油产品运动黏度测定器,乌式黏度计(毛细管内径0.53mm),G3垂熔漏斗,恒温水浴:控温精度±0.05℃,秒表:分度0.01 s,电子天平(精度0.1mg和0.01g),HG53卤素水分测定仪,DHG-9204A型电热恒温鼓风干燥箱。
2 方法
2.1 透明质酸精品的收集
收集1个批次精品透明质酸(批号:HA-T 0812161,山东福瑞达生物医药有限公司生产),放入电热鼓风干燥箱内110℃加热,采用干热法对透明质酸进行降解,按不同的加热时间取样,装入西林瓶中密封,保存至4℃冰箱待用,共得到46份样品。
2.2 透明质酸分子量测定
2.2.1特性黏度法测定透明质酸分子量
采用特性黏度法测定透明质酸得到黏均分子量。结果如表3-1
2.2.1.1供试品溶液
精密称取透明质酸约100 mg至100ml容量瓶中,加0.2mol/L氯化钠溶液适量,置振荡器上使其溶解后稀释至刻度,摇匀。根据测定的供试液的流出时间对上述溶液稀释至合适浓度,摇匀,作为供试液。
2.2.1.2测定
取乌氏黏度计用铬酸浸泡,洗净,调节水浴温度25±0.1℃。
乌氏黏度计用供试液反复冲洗内表面2-3次,自管口倒入供试液,至B球两条刻度线之间,水浴恒温15min,自管口抽气使供试液充满A球,并超过测定线上线,松开管口,供试液在管内自然下落,用秒表计算从上测定线至下测定线时间,重复2次,取均值记为t1。测定溶剂流出时间记为t0。应符合t1/t0=1.3-1.5,t0>100″;溶剂及供试液均经G3垂熔漏斗过滤。
2.2.1.3计算
按公式(3)计算特性黏度:
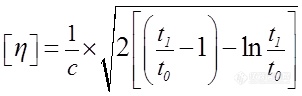 (3)
(3)
式中:t1为供试液的平均流出时间,s(秒)。
t0为溶剂的流出时间,s(秒)。
c为供试液的浓度(按干燥品计算),g/dL。
按公式(4)由特性黏数计算分子量:
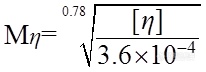 (4)
(4)
2.2.2 多角度激光散射结合凝胶渗透色谱法测定透明质酸分子量
采用多角度激光散射结合凝胶渗透色谱法(GPC-MALLS)测定透明质酸重均分子量。
2.2.2.1色谱分析系统组成
Waters 515单元泵,进样环(200μl),DAWN HELEOS-Ⅱ型多角度激光光散射仪和示差折光检测器(美国Wyatt公司),ASTRA 5.3.4.13软件系统 (美国Wyatt公司)。
2.2.2.2色谱条件
色谱柱:SHODEX 806M型凝胶色谱柱。
流动相:0.2mol/L NaNO3 (含0.02% NaN3),0.22μm滤膜过滤,待用。流速:0.6 ml/min。进样量200μl,dn/dc = 0.160。室温操作。
进样浓度:0.5mg/ml。
2.3 近红外光谱的采集
将透明质酸样品置于自制样品池中,用样品池盖将样品刮平,将样品池放置于支架上,光谱仪的探头卡在样品盒盖的圆孔中,垂直卡紧,采用ratio mode的方式采集样品的近红外光谱,波长范围为1100-2300nm,波长增量2nm,扫描次数为300,每个样品采集2张光谱,求取平均光谱。
2.4 校正集、验证集样品的划分
采用K-S分类方法,对样品集进行划分。
2.5 一级分析方法和建模算法对模型的影响
本实验考察了一级分析方法(特性黏度法和GPC-MALLS法)对模型性能的影响。同时为考察透明质酸分子量与近红外光谱之间的线性关系,本论文分别采用非线性算法SVM和线性算法PLS分别建立透明质酸分子量的校正模型,考察不同的建模算法对模型的影响。对于非线性问题, SVM的主要思想是将原问题变换转化为某个高维空间的线性问题,并在高维空间中进行线性求解。SVM在解决小样本、非线性及高维模式识别中具有许多特有的优势。
3结果
3.1透明质酸分子量的测定结果
采用特性黏度法和GPC-MALLS法测定干热法降解的透明质酸的分子量,测定结果如表1所示。
表1 46个降解透明质酸样品的分子量信息
| 样品 | 黏均分子量 (kDa) | 重均分子量(kDa) | 样品 | 黏均分子量 (kDa) | 重均分子量 (kDa) |
| 1 | 2.722E+03 | 2.314 E+03 | 24 | 1.566E+03 | 1.519 E+03 |
| 2 | 2.582E+03 | 2.433 E+03 | 25 | 1.646E+03 | 1.723 E+03 |
| 3 | 2.567E+03 | 2.243 E+03 | 26 | 1.580E+03 | 1.462 E+03 |
| 4 | 2.442E+03 | 2.262 E+03 | 27 | 1.677E+03 | 1.550 E+03 |
| 5 | 2.478E+03 | 2.274 E+03 | 28 | 1.471E+03 | 1.391 E+03 |
| 6 | 2.418E+03 | 2.152 E+03 | 29 | 1.601E+03 | 1.543 E+03 |
| 7 | 2.339E+03 | 2.14 E+03 | 30 | 1.389E+03 | 1.353 E+03 |
| 8 | 2.196E+03 | 1.803 E+03 | 31 | 1.435E+03 | 1.346 E+03 |
| 9 | 2.277E+03 | 2.100 E+03 | 32 | 1.402E+03 | 1.306 E+03 |
| 10 | 2.080E+03 | 2.065 E+03 | 33 | 1.370E+03 | 1.596 E+03 |
| 11 | 2.097E+03 | 2.293 E+03 | 34 | 1.303E+03 | 1.310 E+03 |
| 12 | 1.967E+03 | 1.793 E+03 | 35 | 1.325E+03 | 1.348 E+03 |
| 13 | 2.134E+03 | 2.001 E+03 | 36 | 1.328E+03 | 1.392 E+03 |
| 14 | 1.929E+03 | 1.931 E+03 | 37 | 1.299E+03 | 1.376 E+03 |
| 15 | 1.946E+03 | 1.854 E+03 | 38 | 1.255E+03 | 1.247 E+03 |
| 16 | 1.855E+03 | 1.757 E+03 | 39 | 1.244E+03 | 1.217 E+03 |
| 17 | 1.917E+03 | 1.782 E+03 | 40 | 1.205E+03 | 1.274 E+03 |
| 18 | 1.756E+03 | 1.647 E+03 | 41 | 1.166E+03 | 1.062 E+03 |
| 19 | 1.868E+03 | 1.840 E+03 | 42 | 1.160E+03 | 1.051 E+03 |
| 20 | 1.716E+03 | 1.600E+03 | 43 | 1.083E+03 | 9.563 E+02 |
| 21 | 1.809E+03 | 1.577 E+03 | 44 | 1.096E+03 | 1.065 E+03 |
| 22 | 1.633E+03 | 1.502 E+03 | 45 | 1.100E+03 | 9.820 E+02 |
| 23 | 1.809E+03 | 1.548 E+03 | 46 | 2.710E+03 | 2.595 E+03 |
可以看出,得到的透明质酸黏均分子量和重均分子量的范围分别为1100kDa-2722 kDa和956.3kDa-2595kDa,具有一定的范围和梯度,适合用于建立近红外模型。
3.2 透明质酸精品的近红外光谱
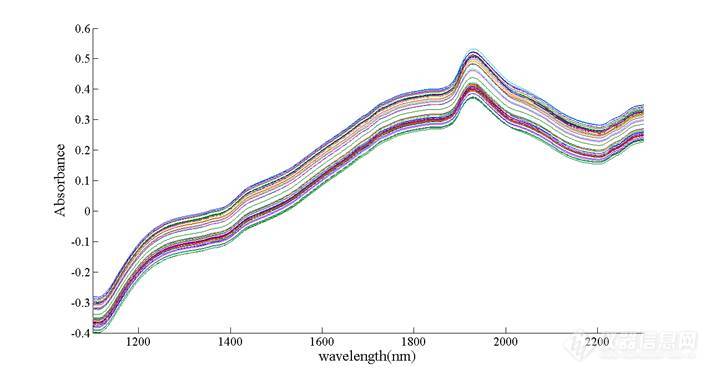
图1 46个透明质酸样品的近红外漫反射光谱图
图1显示了46份透明质酸固体精品的原始漫反射光谱。由于透明质酸具有很强的吸湿性,在 1400nm和1900nm附近的光谱分别为水的存在所产生的吸收。因此,在以下的处理中,去掉1365-1450nm,1850-1950nm波段。
3.3 校正集、验证集样品的划分结果
采用K-S分类方法将样品集进行划分,其中30个样品为校正集样品、16个样品为验证集样品。
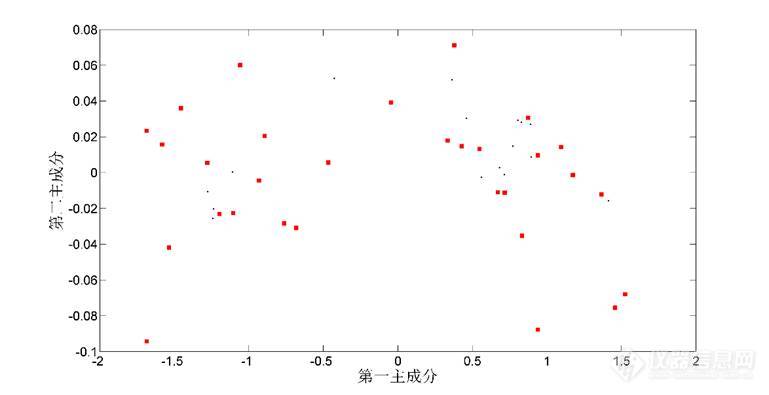
图2 46个透明质酸样品的主成分得分图
注:红色方块为校正集样品;黑色星号为验证集样品
图2为通过K-S法挑选的46个样品的主成分得分图。可见30个校正集样品在主成分图上分布较均匀,而且16个验证集样本也都被包容在校正集的空间内,这说明验证集的光谱信息被包容在校正集的光谱信息内,符合理想校正集和验证集的要求,通过上述方法可以提高模型预测精度。
样品集划分信息如表2和表3所示。
表 2 样品集信息(黏均分子量)
| 样品集 | 数量 | 最大值 (kDa) | 最小值 (kDa) | 平均值 (kDa) | 标准偏差 |
| 校正集 | 30 | 2.722E+03 | 1.083E+03 | 1.815E+03 | 4.966E+02 |
| 验证集 | 16 | 2.582E+03 | 1.096E+03 | 1.655E+03 | 4.374E+02 |
表 3 样品集信息(重均分子量)
| 样品集 | 数量 | 最大值 (kDa) | 最小值 (kDa) | 平均值 (kDa) | 标准偏差 |
| 校正集 | 30 | 2.592E+03 | 9.563E+02 | 1.699E+03 | 4.250E+02 |
| 验证集 | 16 | 2.433E+03 | 9.820E+02 | 1.600E+03 | 4.201E+02 |
3.4一级分析方法和建模算法对模型的影响
对透明质酸的黏均分子量和重均分子量,分别采用SVM和PLS方法建立透明质酸分子量的校正模型,考察不同的一级分析方法和建模算法对模型准确度和精密度的影响。本实验分别采用MSC、SNV、一阶导数9点平滑3种预处理方法对原始光谱进行预处理,在去水后的1100-1364nm、1452-1848nm、1952-2300nm范围内,建立透明质酸黏均分子量和重均分子量的SVM分析模型和PLS分析模型。
采用SVM方法分别建立黏均分子量和重均分子量模型,采用交互验证和网格寻优的方法选择最佳的惩罚系数和核函数参数,交互验证结果见图3,并采用建立的SVM模型对验证集的样品进行预测,预测结果见图4。
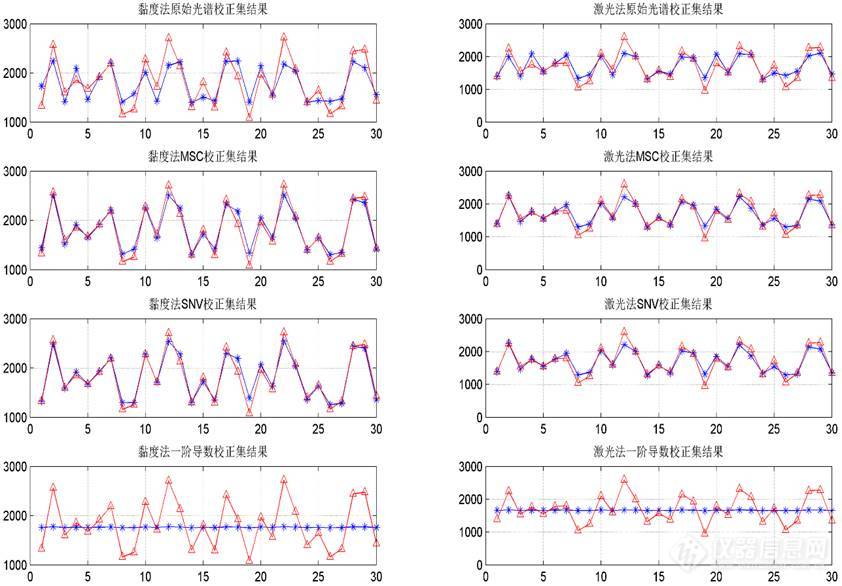
图3 SVM模型校正集预测结果图
注:红色三角表示样品真实值;蓝色星号为样品集预测值
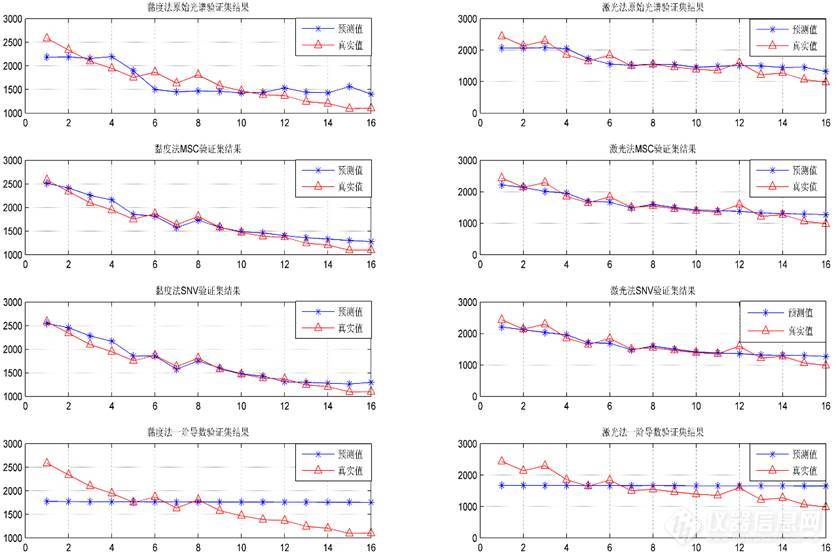
图4 SVM模型验证集预测结果图
注:红色三角表示样品真实值;蓝色星号为样品集预测值
表4为不同的预处理方法下采用SVM方法建立的黏均分子量和重均分子量模型的各相关参数。
表4 SVM模型各相关参数
| 一级数据来源 | 预处理方法 | 惩罚系数 | 核函数参数 | Rc2 | RMSECV | Rp2 | RMSEP |
| 特性黏度法 | 原始光谱 | 256 | 0.3535 | 0.7422 | 307 | 0.6608 | 249 |
| 特性黏度法 | 一阶导数 | 256 | 256 | 0.9552 | 493 | 0.9614 | 432 |
| 特性黏度法 | MSC | 256 | 45.25 | 0.9592 | 154 | 0.9506 | 115 |
| 特性黏度法 | SNV | 256 | 4 | 0.9569 | 158 | 0.9526 | 110 |
| GPC-MALLS | 原始光谱 | 256 | 4 | 0.7801 | 265 | 0.8173 | 213 |
| GPC-MALLS | 一阶导数 | 256 | 256 | 0.9285 | 416 | 0.9076 | 407 |
| GPC-MALLS | MSC | 256 | 32 | 0.9180 | 152 | 0.9002 | 48 |
| GPC-MALLS | SNV | 256 | 1.442 | 0.9187 | 158 | 0.8982 | 152 |
表5 PLS模型各相关参数
| 一级数据来源 | 预处理方法 | Rc | RMSECV | Rp | RMSEP | PCs |
| 特性黏度法 | 原始光谱 | 0.9823 | 91.77 | 0.9946 | 67.31 | 3 |
| 特性黏度法 | 一阶9点 | 0.9832 | 88.39 | 0.9913 | 69.41 | 2 |
| 特性黏度法 | MSC | 0.9847 | 85.15 | 0.9951 | 67.89 | 3 |
| 特性黏度法 | SNV | 0.9747 | 109.2 | 0.9839 | 97.94 | 1 |
| GPC-MALLS | 原始光谱 | 0.9593 | 118.2 | 0.9532 | 124.8 | 3 |
| GPC-MALLS | 一阶9点 | 0.9550 | 122.6 | 0.9598 | 124.0 | 1 |
| GPC-MALLS | MSC | 0.9636 | 111.9 | 0.9574 | 118.0 | 3 |
| GPC-MALLS | SNV | 0.9587 | 118.8 | 0.9560 | 121.4 | 1 |
从表4和表5可知,特性黏度法测得的一级数据所建立的黏均分子量的校正模型,准确度和精密度均好于GPC-MALLS法测得的一级数据建立的重均分子量模型。
4 讨论
本实验采用两种不同的一级分析数据对透明质酸分子量进行近红外建模。黏均分子量所建立模型优于重均分子量,这可能与分子量的测定机理有关系。黏均分子量是分子量对分子链流体力学体积的算术平方根的统计平均,而重均分子量是各种分子量的分子的重量分数与其相应的分子量的乘积,即对分子链重量的统计平均,在统计意义上要比黏均分子量的线性关系弱。此外,还与一级分析方法本身的误差有关。同时,无论采用何种预处理方法,PLS算法建立的模型均好于SVM算法所建立的模型。由于SVM算法是一种非线性的学习方法,PLS为线性的多元校正方法,因此可以说明无论是黏均分子量还是重均分子量均与透明质酸近红外光谱线性关系更强,适合建立线性模型。
参考文献
Laurent T C, Gergly J. Light-scattering studieson hyaluronic acid. J. Bio.l Chem.,1955, 212(1):325-333.
Gleland R L, Wang J L. Ionic polysaccharides. Ⅲ. Dilute solution properties of hyaluronic acidfractions. Biopolymers, 1970,9(7):799-810.
杨桂兰, 郭学平, 栾贻宏. 不同相对分子质量透明质酸钠的应用. 食品与药品, 2005, 7(12): 1-3.
Li M,Rosenfeld L, Vilar R E, et al.Degradation of hyaluronan by peroxynitrite. Arch. Biochem. Biophys., 1997, 341(2): 245-250.
SoltesL, Mendichi R, Lath D, et al.Molecular characteristics of some commercial high-molecular-weight hyaluronans. Biomed. Chromatogr., 2002, 16(7): 459-462.
Terbojevich M, Cosani A, Palumbo M. Molecular weight distribution ofhyaluronic acid by high-performance gel permeation chromatography. Carbohydr. Res., 1986, 157: 269-272.
KarlssonG, Bergman R. Determination of the distribution of molecular masses of sodiumhyaluronate by high-performance anion-exchange chromatography. J. Chromatogr. A., 2003, 986(1): 67-72.
丁厚强,王海英,郭学平.多角度激光光散射仪与尺寸排阻色谱法联用测定透明质酸相对分子质量及其分布. 食品与药品. 2009, 11(3): 24-26.
WahlundK G, Litzén A. Application of an asymmetrical flow field-flow fractionationchannel to the separation and characterization of proteins, plasmids, plasmidfragments, polysaccharides and unicellular algae. J. Chromatogr. A., 1989, 461: 73-87.
TakahashiR, Al-assaf S, Williams P A, et al.Asymmetrical-flow field-flow fractionation with on-line multiangle lightscattering detection. 1. Application to wormlike chain analysis of weakly stiffpolymer chains. Biomacromolecules,2003, 4(2): 404-409.
HayaseS, Oda Y, Honda S, et al.High-performance capillary electrophoresis of hyaluronic acid: determination ofits amount and molecular mass. J.Chromatogr. A., 1997, 768(2): 295-305.
KinoshitaM, Shiraishi H, Muranushi C, et al.Determination of molecular mass of acidic polysaccharides by capillaryelectrophoresis. Biomed. Chromatogr.2002, 16(2): 141-145.
Lee H G,Cowman M K. An agarose gel electrophoretic method for analysis of hyaluronanmolecular weight distribution. Anal.Biochem., 1994, 219(2): 278-287.
ArmstrongS, Bell D R. Measurement of high-molecular-weight hyaluronan in solid tissueusing agarose gel electrophoresis. Anal.Biochem., 2002, 308(2): 255-264.
Hokputsa S, Jumel K, Alexander C, et al. A comparison of molecular massdetermination of hyaluronic acid using SEC/MALLS and sedimentationequilibrium. Eur. Biophys. J.,2003, 32(5): 450.
TakagakiK, Kojima K, Majima M, et al.Ion-spray mass spectrometric analysis of glycosaminoglycan oligosaccharides.Glycoconj. J., 1992, 9(4): 174.
Yeung B,Marecak D. Molecular weight determination of hyaluronic acid by gel filtrationchromatography coupled to matrix-assisted laser desorption ionization massspectrometry. J. Chromatogr. A., 1999,852(2): 573



































