

实验条件改变对实验结果的影响
一次惊险的误判
实验室收到一批食用油样品,需要做其中的苯并【α】芘,经过前期的实验摸索,现在对这个实验已经驾轻就熟(详见帖子:http://bbs.instrument.com.cn/topic/6025769?UID=vcningmeng&sortby=desc),经过一系列的实验及方法验证,后续又在原来的实验方法基础上进行了改动。

针对食用油样品,改动后的样品前处理方法如下:
准确称取约0.5g样品于5mL刻度试管中,加入乙腈并定容至刻度,漩涡混匀,利用乙腈提取样品中的苯并【α】芘。混匀的样品经高速离心机离心,取上清液过0.22μm滤膜后进样并上机检测。该检测方法与国标方法相比减少了固相萃取柱使用量,缩短样品前处理时间,同时能够极大提高样品检测的效率。
色谱条件如下:
色谱柱:C18 (250mm×4.6mm ,5μm);
流动相:甲醇:水=95:5;
流速:1.0mL/min;
进样体积:20μL;
柱温:35℃;
荧光检测器:激发波长384nm,发射波长406nm
标准工作溶液及样品测定的色谱图如下:
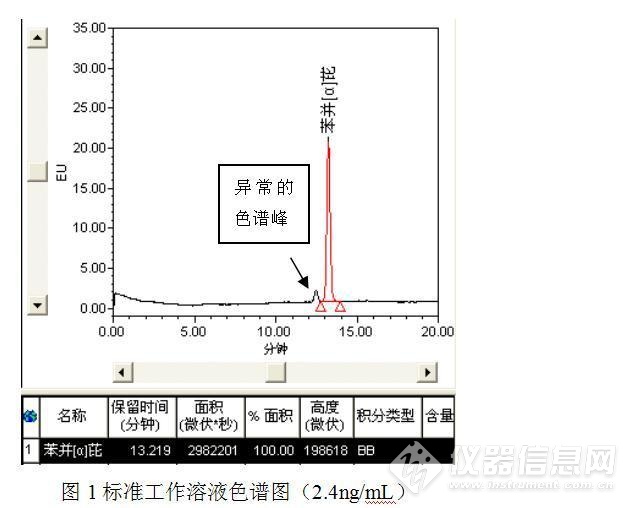
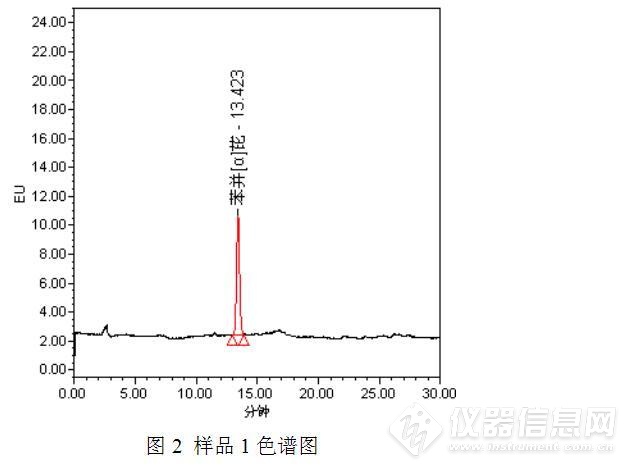
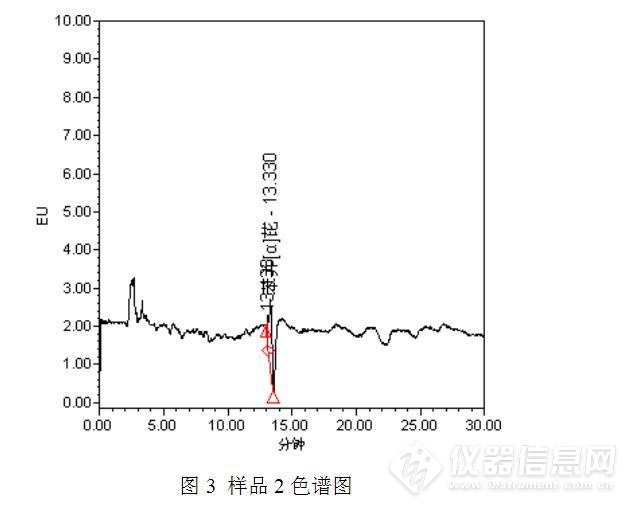
经过测试发现,与之前的测试结果相比,发现了3点异常之处:(1)保存于冰箱中的苯并【α】芘储备液可能发生了降解或者被污染了,主峰附近出现了一个小的色谱峰;(2)部分样品(如样品2)在苯并【α】芘保留时间附近出现了倒峰,影响了待测物质苯并【α】芘的测定;(3)此次测定的食用油中均检出了苯并【α】芘,而且从峰高初步判断,苯并【α】芘的含量均较高,异于平常的测定值。
针对问题1:储备液发生异常,于是重新配制标准工作溶液,但是在测试过程中又发现了新的问题,由于更换了供应商,新采购的标准溶液稀释后的测定值与原先相比低了许多,新配制的标准工作溶液如图4。
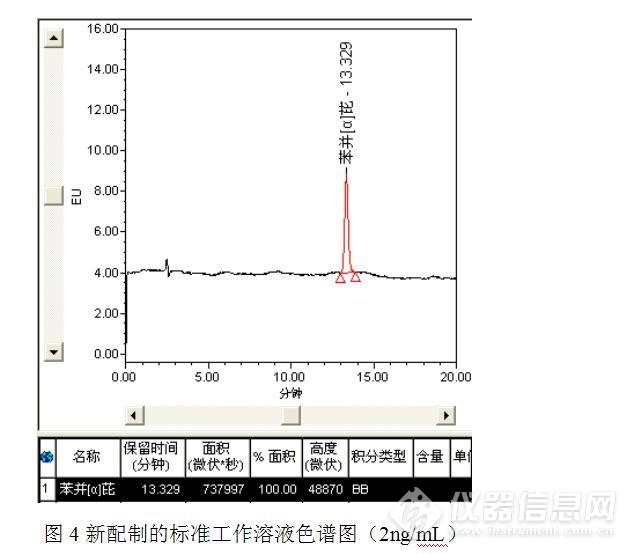
更换人员、更换溶剂,经过多次配制测试发现,新购标准溶液稀释后2ng/mL标准工作溶液的峰面积稳定在70万微伏*秒,排除了人员失误、溶剂效应和单位换算等问题,具体原因还在于对方协商、讨论之中。尽管峰面积有异常,但是原先主峰附近的异常的色谱峰消失了。
针对问题2:倒峰影响了待测物质苯并【α】芘的测定,于是想优化色谱条件,看能否将两者分开。又考虑问题1,待测物质苯并【α】芘峰面积变小,是否与流动相中有机相的更换有关,毕竟甲醇的极性大于乙腈,在反相色谱中乙腈的洗脱能力要强于甲醇的。于是将甲醇更换为乙腈并重新测定,将原先的流动相甲醇:水=95:5更换为乙腈:水=95:5,测定结果见图5~图7。
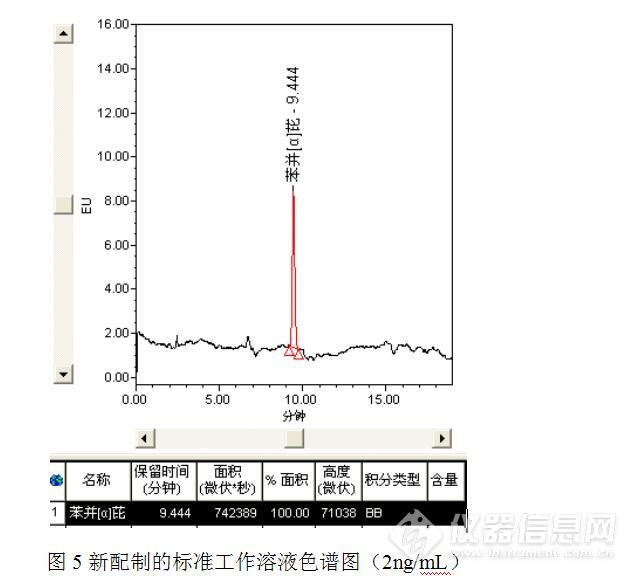
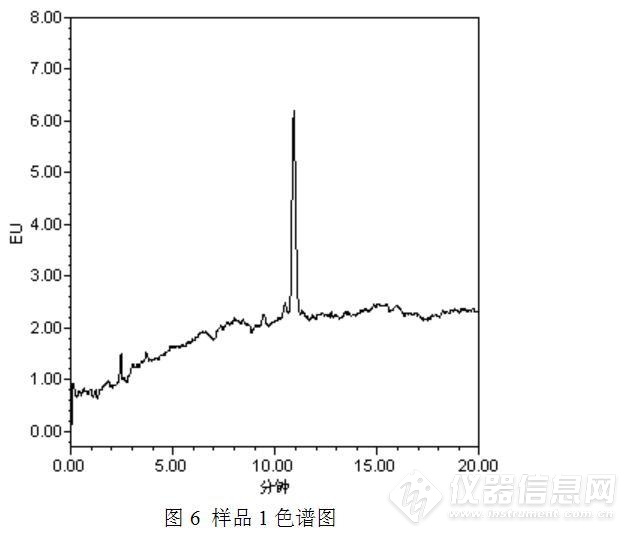
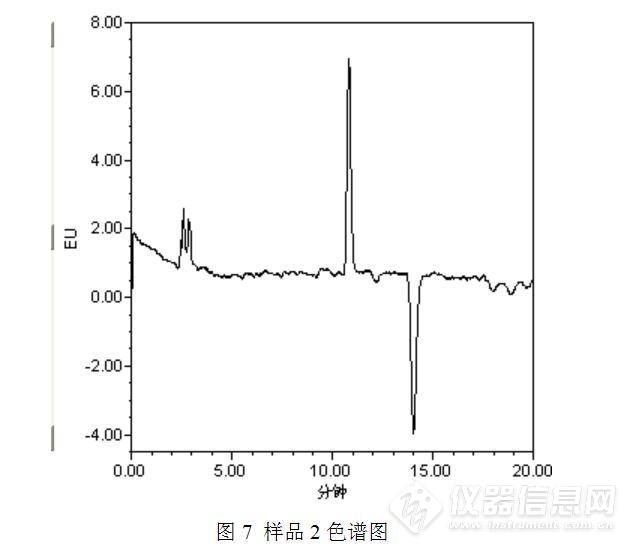
测试结果表明,将流动相甲醇:水=95:5更换为乙腈:水=95:5,对苯并【α】芘色谱峰面积的影响并不大,但是流动相改变之后,苯并【α】芘的保留时间发生了变化,从而与原先的倒峰发生了分离,消除了倒峰对苯并【α】芘测定的影响。但是随之又产生了一个新的问题,以乙腈:水=95:5为流动相时,样品中苯并【α】芘与标准工作溶液中苯并芘的保留时间发生了偏移。初步估计是样品基质的影响,食用油是油性样品,提取液乙腈是极性溶剂,两者不相溶,混匀、离心后的乙腈中可能会存在微小的油性小液滴,从而导致保留时间的漂移。为了验证这一假设,按照食品安全国家标准GB 5009.27 -2016中采用苯并【α】芘分子印迹柱方法进行前处理,因为该方法通过苯并【α】芘分子印迹柱实现了浓缩、富集、净化及溶剂替换的过程,具体前处理方法如下:
(1)称取0.5g试样,加入5mL正己烷,旋涡混合0.5min,待净化;
(2)采用苯并【α】芘分子印迹柱,依次用5mL二氯甲烷及5mL正己烷活化柱子。将待净化液转移进柱子,待液面降至柱床时,用6mL正己烷淋洗柱子,弃去流出液。用6mL二氯甲烷洗脱并收集净化液到试管中。将净化液在40℃下氮气吹干,准确吸取1mL乙腈涡旋复溶0.5min,过0.22μm微孔滤膜后供液相色谱测定,测定结果见图8.
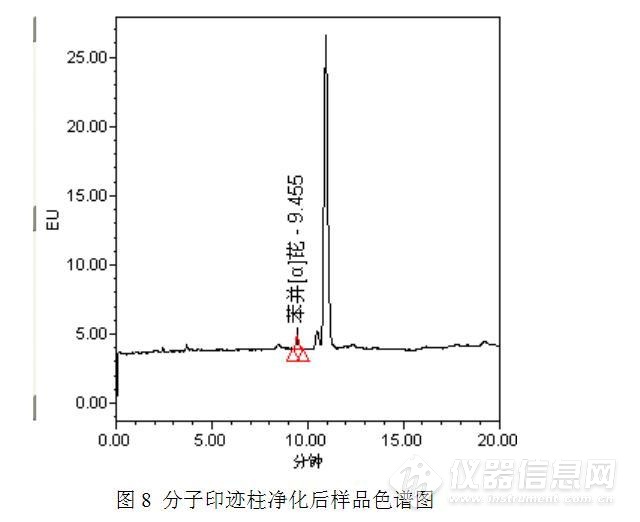
测试结果表明,经过分子印迹柱净化后11min左右的色谱峰依然存在,与此同时9.4min附近出现了一个苯并【α】芘的小色谱峰,该色谱峰一方面与溶剂的本底有关(之前的帖子有讨论),另一方面是分子印迹柱富集、浓缩的结果。苯并【α】芘色谱峰的出现以及11min左右的色谱峰保留时间没有改变,初步判定11min左右的色谱峰为干扰峰。为了验证这一想法,往样品瓶中加入标准工作溶液,进行简单的加标定性验证,加标测试结果见图9。

通过加标实验,进一步确定了11min左右的色谱峰非苯并【α】芘的色谱峰,这时倒为开始的误判捏了一把冷汗,幸亏倒峰的出现,才成功的避开了一次失误。
接下来的实验,纯属个人好奇心太重,尝试测定一下11min左右的色谱峰是何种物质。
首先进行抗氧化剂的测定,以乙腈:水=95:5为流动相,进了一针没食子酸丙醋(PG)、叔丁基对苯二酚(TBHQ)、叔丁基对经基茵香醚(BHA)和2,6一二叔丁基对甲基苯酚(BHT )的4种抗氧化剂的混合标准工作溶液,测定结果见图10。
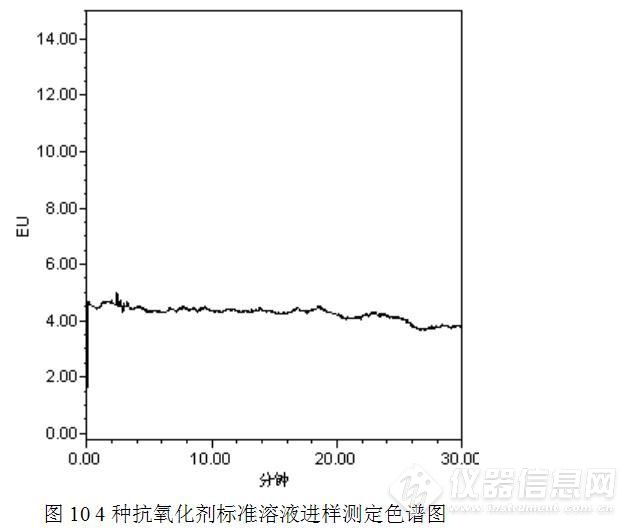
测定结果表明,实验中用到的4种抗氧化剂在该色谱条件下并没有响应。
接着测定黄曲霉毒素B1/B2/G1/G2,测定方法参考食品安全国家标准GB 5009.22-2016,测定结果见图11和图12.
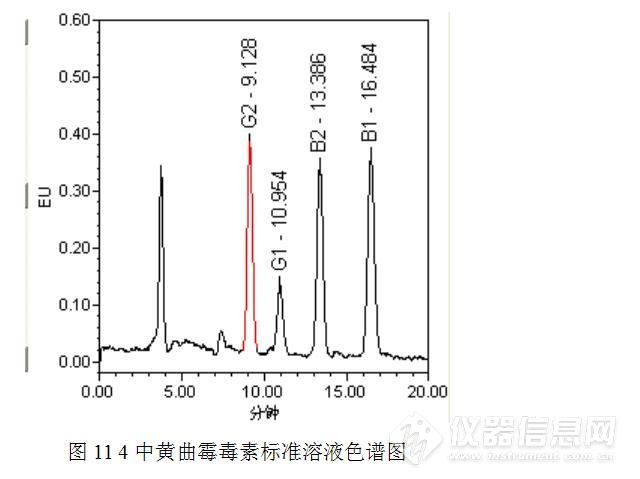
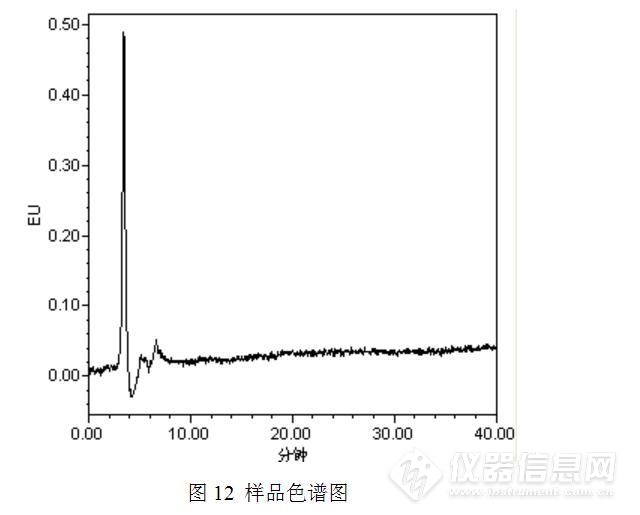
经过测试,排除了11min左右的色谱峰是上述4中黄曲霉毒素的可能性。受限于标准品和仪器设备,没有进行下一步的实验,有兴趣的实验猿可以进一步尝试,或许有新的发现。
后记:在后续的交流沟通工作中,对方提供了苯并【α】芘固态标准品,然后经过溶解、稀释后测定,在相同浓度下,固态标准品稀释后测定的结果与新购买的标准工作溶液测定结果一致,貌似说明了一些问题。同时也提出了一个问题,在初次实验或者方法开发过程中,如何应对或避免标准品或标准溶液变质、浓度及含量不准确对后续实验造成的影响。



 。检测的盲区,有质谱还能质谱分析一下
。检测的盲区,有质谱还能质谱分析一下































