

miaoweili 2007/08/21
流动相极性调大一点
ljd5210 2007/08/29
用岛津的UFLC,超快速高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url][em17]
wubaicang2003 2007/08/18
把水重新蒸馏一下,再重配流动相;在正式做样之前最好不进样就走一个空梯度让系统充分饱和,设定梯度时最后让流动相配比回归到起始比例,你的问题应该能够解决
yaya91 2007/08/20
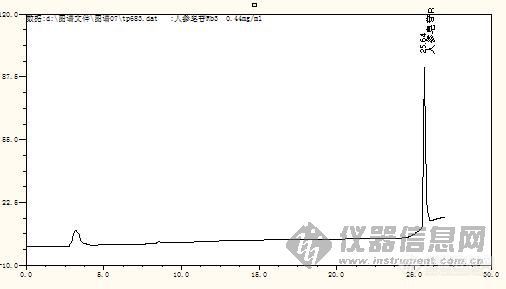
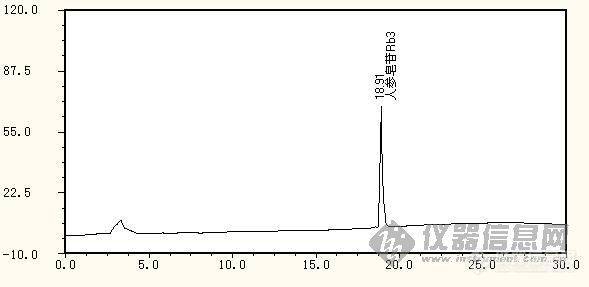
只要分离度达到1.5以上,就可以了.从图谱上看,样品的主成分峰峰形与对照品一致,条件是合适的。假如峰提前,会导致与相关杂质分不开。
Easy-Boy 2007/08/21
[quote]原文由 [B]yaya91[/B] 发表: 只要分离度达到1.5以上,就可以了.从图谱上看,样品的主成分峰峰形与对照品一致,条件是合适的。假如峰提前,会导致与相关杂质分不开。[/quote] 是啊,你可以用溶剂走一个空白,然后减去空白图谱,就能够获得基线较平整的图谱了。如果是Waters的仪器。
tanggangfeng
第3楼2007/08/20
[quote]原文由 txm 发表:2005版《中国药典》采用HPLC法测定了人参皂苷Rb3的含量,色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂。以乙腈为流动相A,以0。2%磷酸溶液为流动相B,按下式进行梯度洗脱,检测波长为203nm。...
0~19分钟:流动相A(%):30→35;流动相B(%):70→65
19~21分钟:流动相A(%):35→60;流动相B(%):65→50
21~26分钟:流动相A(%):50;流动相B(%):50
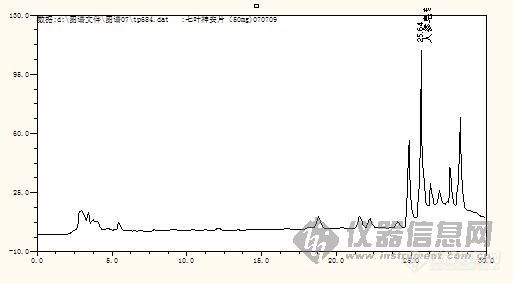
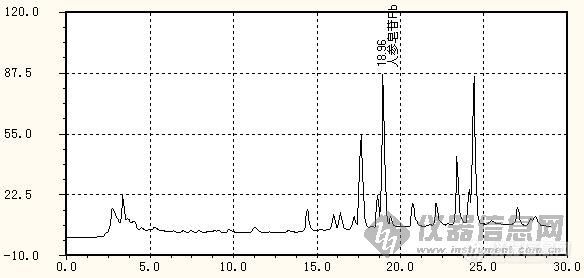
从图谱上看,应该还有优化可能。前面近20分钟基本没出峰。
可以尝试改下,如
0~10分钟:流动相A(%):30→35;流动相B(%):70→65
10~26分钟:流动相A(%):35→60;流动相B(%):65→40
txm
第7楼2007/08/22
首先非常感谢大家对我提出的问题热心解答!
我来总结一下:
1.把水重新蒸馏一下,再重配流动相;在正式做样之前最好不进样就走一个空梯度让系统充分饱和,设定梯度时最后让流动相配比回归到起始比例。
这个方法我在任何梯度实验时都是这样做的,而且来回重复做了几次,换了两根进口柱子,结果没有什么改变。
2.只要分离度达到1.5以上,就可以了.
分离度达到1.5,我们还要求峰走在一条直的基线上,而且主成分峰出现了一些锯齿峰,因图被缩小了,所以在图上看不出来。
3.由tanggangfeng提供的梯度洗脱比例,这个可以试一下,等待看结果如何再告知大家!!!
最后还是非常感谢大家如此热心提供方法!!!!!

































