推荐厂家
暂无
暂无






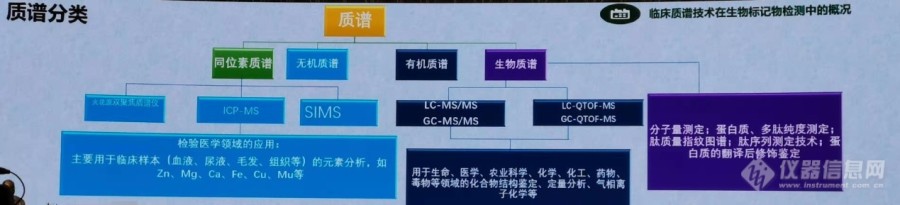
[align=center][b]临床质谱为精准医学保驾护航[/b][/align]精准医学,Precision Medicine 是一种将个人基因、环境与生活习惯差异考虑在内的疾病预防与处置的新兴方法。国外精准医学的历史:2011年,美国医学界首次提出“精准医学”的概念;2015年1月20日,奥巴马在美国国情咨文中提出“精准医学计划”,希望精准医学可以引领一个医学新时代。国内精准医学的历史:2006年中国首次提出“精准外科”的概念;2015年首届精准医疗战略专家委员会在上海成立;2016年精准医疗首次进入政协提案。短短的15年间,检验技术由生化发光检测生化项目、发光产品,革新至五年前的分子诊断检测基因测序、基因诊断,自2018年元年,2019年起始年,质谱技术发展得到井喷式爆发。现代精准医学的研究模式:测序基因组的同时,搜集所有表型信息,将基因与表型大数据结合起来。诊断模式主要有个人基因组信息、蛋白质组学和代谢组学,其中蛋白质组学和代谢组学需要临床质谱“大显神威”。治疗方式有针对基因组进行个人用药与药物设计、药物代谢和毒理评估。治疗效果:医疗资源耗费降低,针对性用药提升疗效,药物副作用降低。质谱技术在医学检测应用中的发展:1981年美国Nimitz航母事件促使医学对质谱仪器的需求,1988年美国联邦药品检验局发布强制性指南,要求治疗药物必须使用质谱法进行确认。1990年,MS开始用于新生儿筛查,1996年,GC-MS用于解决睾酮免疫分析问题,同年MS用于完整细菌的快速鉴定。1998年,GC-MS和[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]开始用于临床检验中。2004年,MS用于鉴别血源性感染中的化合物,2010年,国内首家质谱临床检验试验室成立,2010年质谱用于代谢组学、蛋白质组学和其它组学。2013年,MS用于类固醇化合物鉴定的研究论文被大量发表,同年FDA首次批准MALDI-TOF用于微生物的鉴定。2016年实时MS技术引导下的肿瘤手术刀出现。而临床质谱在国际影响力也越来越大,1906年,J.L.Thomson 获得诺贝尔物理学奖,他发现由电子组成“阴极射线”,并测量了电子的荷-质比。1989年,W.Paul 获得诺贝尔物理学奖,他的贡献是发明了离子肼技术。2002年,J.B.Fenn和田中耕一获得诺贝尔化学奖,他们分别发现了电喷雾ESI电离方法生物大分子分析、基质辅助激光解吸电离质谱MALDI电离方法生物大分析分析。质谱仪发展至今已出现多个分支,下图是质谱分类。[img=,690,157]https://ng1.17img.cn/bbsfiles/images/2019/08/201908132201538393_4418_3255306_3.jpg!w690x157.jpg[/img]在我国临床质谱的发展态势明显,相关的政策法规如下:2016年3月国家卫计委为临床实验室自建项目(LDT)开启绿色通道,临床检验进入新发展时期。2018年2月1日,我国质谱行业首个通用规范《质谱仪通用规范》实施,该国标将引领质谱行业规范健康发展。今年健康强国战略深入推进,第三方检测机构快速发展,越来越多医学实验室加大投入更大规模的质谱平台建设。但质谱仪在大规模使用方面还有很多局限。我想主要是以下几个方面制约了质谱仪进一步大规模发展使用:1、缺乏罗西贝雅这样整合仪器试剂产品的整体解决方案提供者。质谱仪均由国外仪器厂家提供,而试剂由试剂厂商提供,仪器试剂分属不同的而厂家。推广力度也存在问题,售后问题难以解决。2、样品前处理步骤繁琐:以维生素项目为例,前处理一共涉及到13步操作(萃取、孵育、振荡、离心、氮吹等等),且数据波动大,可靠性低。3、仪器操作复杂,按照现有生化、化学发光技术几天的培训力度,医院技术人员无法熟练操作质谱仪。4、使用项目少,只有新生儿遗传代谢病的检测,综合三甲医院买了仪器无法高效的使用。但综合我国临床质谱的应用现例,我们可以发现还是有很多可圈可点的。例如,新生儿筛查,克服了传统新生儿筛查传统分析方法的缺点,一种实验检测一种疾病,工作量随着样本数大大增加等。[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS凭借其高通量、高灵敏度,可以一次分析检验多种疾病,且假阳性率低,筛查效率高,结果可靠,综合费用相对低廉,检测速度很快,一般一个样品在2-3分钟。在甲氨蝶呤的检测中,质谱仪发挥其巨大潜能。传统的FPIA法Abbott TDx-FLx药物浓度分析仪和EMIT法西门子Viva-E全自动药物浓度检测系统均具有缺点,如设备不再更新、测定MTX时存在正偏差等。[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS可将药物与代谢产物以及内源性物质分离,具有专一性强的特点,是检测MTX血浆浓度的金标准,但改法仍需要复杂的前处理过程现如今,质谱技术应用仍存在难点:质谱人才匮乏、基于质谱方法开发和优化的复杂程度较难、质谱方法性能的验证复杂、成本较高等。如何利用好这一未来前景巨大的技术是每个医院、药企需要考虑的问题,相信在未来的研究中,质谱仪将继续表现巨大的应用前景!
[align=center][b][font=宋体][color=black]脑卒中疾病个体化精准用药[/color][/font][/b][/align][align=left][/align][align=left][/align][b][/b][align=left][b][font=宋体][color=black]摘要:[/color][/font][/b][/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]提供一种指导脑卒中疾病个体化精准用药的系统及方法。该系统采用药物基因组检测技术,获得个体药物相关基因组的基因型;然后根据大数据基础上建立的药物基因组数据库,结合已有的临床用药指南或共识,预测疗效、预警副作用,制定初步的个体化精准用药方案。然后在临床用药过程中,进行药物浓度监测,获得个体体内实际的药物浓度,结合临床症状改善情况、不良反应发生情况,优化调整药物的种类、剂量、频次、给药途径等,以在特定患者和特定疾病正确诊断的基础上,在正确的时间、给与正确的药物、使用正确的剂量,实现真正的个体化精准用药。[/align][font='微软雅黑',sans-serif][/font][align=left][/align][b][/b][align=left][b][font=宋体][color=black]背景技术[/color][/font][font=宋体][color=black]:[/color][/font][/b][/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left][font=宋体][color=black]脑卒中,即民间俗称的[/color][/font][font=宋体][color=black]“羊角风”或“羊癫风”,是临床诊疗中常见的一种疾病,主要是患者因各种原因导致大脑神经元突然异常放电,引发大脑神经功能暂时紊乱所致,一般需要及时进行针对性的药物治疗,以缓解症状,克服并发症。据相关统计,我国脑卒中疾病的发病率在7%左右。脑卒中疾病的发病与脑部疾病、遗传因素、全身性的系统疾病的发生等有一定的关系。目前, 老年人脑卒中的发病率居神经系统疾病的第三位,仅在脑血管病、痴呆之后。但因老年人的记忆力和认知功能减退、自诉能力差、老年人脑卒中的表现多不典型,以致常难以被发现而延误诊治。而在脑卒中临床治疗过程中发现,不同的脑卒中疾病患者对于药物治疗存在明显差异。[/color][/font][/align][font=宋体][/font][align=left][/align][align=left] 精准医疗又叫个性化医疗,是指以个人基因组信息为基础,结合蛋白质组,代谢组等相关内环境信息,为病人量身设计出最佳治疗方案,以期达到治疗效果最大化和副作用最小化的一门定制医疗模式。一种指导脑卒中疾病个体化精准用药的系统,可以基于个体的遗传信息预测药物治疗反应,以提高药物的治疗疗效,同时减少毒副作用,实现精准用药。该系统采用药物基因组检测技术,获得个体药物相关基因组的基因型;然后根据大数据基础上建立的药物基因组数据库,结合已有的临床用药指南或共识,预测疗效、预警副作用,制定初步的个体化精准用药方案。在临床用药过程中,进行药物浓度监测,获得个体体内实际的药物浓度,结合临床症状改善情况、不良反应发生情况,优化调整药物的种类、剂量、频次、给药途径等,以期达到精准医疗的目标。[/align][font='微软雅黑',sans-serif][/font][align=left][/align][b][/b][align=left][b][font=宋体][color=black]内容[/color][/font][/b][/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]由于药物的吸收代谢存在个体差异,在临床中需要个体化治疗方案,包括药物种类、治疗剂量,从而减少药效不佳或者严重不良反应等情况。针对以上需求,本专利的目的是建立一种指导脑卒中疾病个体化精准用药的系统,结合药物基因检测、药物浓度监测技术和已有的临床用药指导原则,设计一个合理的个体化用药方案和详细信息,为临床医生合理化用药提供依据,从而解决个体化治疗过程中具体方案制定的问题,包括药物种类、剂量、用药时间和给药途径的选择,使医生快速、准确地对忠者进行用药治疗。[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]为达到上述目的,提供如下技术方案:[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]第一目的在于提供一种指导脑卒中疾病个体化精准用药的系统,该系统包括患者信息模块、药物基因检测模块、数据库模块、初步方案制定模块和报告模块:[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]所述患者信息模块用于记录患者的基本信息;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]所述药物基因检测模块用于检测患者的抗脑卒中药物相关基因的多态性信息;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]所述数据库模块用于储存治疗脑卒中疾病不同候选药物的临床使用信息;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]所述初步方案制定模块,用于利用从患者信息模块、药物基因检测模块和数据库模块导入的信息,分析预估各种治疗方案对该患者个体的预期药效和不良反应风险,进一步判断候选药物与患者是否匹配,并确定患者的初步治疗方案:所述初步治疗方案包括具体药物治疗方案、预期药效和不良反应风险;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]所述报告模块用于生成初步治疗方案报告。[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]第二目的在于提供一种指导脑卒中疾病个体化精准用药的方法,包括:[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]采集患者血浆样本,通过药物基因检测模块检测患者的抗脑卒中药物相关基因多态性信息;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]将药物基因检测模块中检测得到的数据、患者信息模块中的数据、数据库模块中的治疗脑卒中疾病不同候选药物的临床使用信息导入初步方案制定模块;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]初步方案制定模块根据导入的信息,分析预估各种治疗方案对该患者个体的预期药效和不良反应风险,进一步判断候选药物与患者是否匹配,并确定患者的初步治疗方案,所述初步治疗方案包括具体药物治疗方案、预期药效和不良反应风险。[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]第三目的在于提供一种指导脑卒中疾病个体化精准用药的系统,该系统包括:治疗信息更新模块、药物浓度监测模块、数据库模块、方案优化调整模块和报告模块;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]治疗信息更新模块,用于导入初步治疗方案和患者接受初步治疗后的复查结果;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]药物浓度监测模块,用于从患者样本中获得患者个体对抗脑卒中药物的体内暴露水平或代谢水平信息;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]数据库模块,用于储存治疗药物的临床使用数据、临床药物药代动力学研究数据;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]方案优化调整模块,用于利用从治疗信息更新模块、药物浓度监测模块、数据库模块导入的信息,根据患者复查结果以及实际药物暴露水平或代谢水平,综合分析评估初步治疗方案的治疗效果,并进一步对其优化调整,得到优化治疗方案;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]报告模块,用于导出优化调整后的治疗方案报告。[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]第四目的在于提供一种指导脑卒中疾病个体化精准用药的方法,包括:[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]将初步治疗方案和患者采用初步方案治疗后的最新复查结果导入治疗信息更新模块;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]采集患者血浆样本,通过药物浓度监测模块监测患者个体对抗脑卒中药物的体内暴露水平或代谢水平信息;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left][font=宋体][color=black]将治疗信息更新模块中的信息、药物浓度监测模块监测得到的信息、数据库模块中的治疗[/color][/font][font='Times New Roman',serif][color=black]-[/color][/font][font=宋体][color=black]药物的临床使用信息及临床药物药代动力学研究数据,导入方案优化调整模块,方案优化调整模块根据导入的信息,根据患者复查结果以及实际药物暴露水平或代谢水平,综合分析评估初步治疗方案的治疗效果,并进一步对其优化调整,得到优化治疗方案。[/color][/font][/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]第五目的在于提供一种指导脑卒中疾病个体化精准用药的系统,该系统包括患者信息模块、药物基因检测模块、数据库模块、初步方案制定模块、治疗信息更新模块、药物浓度监测模块、方案优化调整模块:[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]所述患者信息模块用于记录患者的基本信息;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]所述药物基因检测模块用于检测患者的抗脑卒中药物相关基因多态性信息;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]所述数据库模块用于储存治疗药物的临床使用信息、临床药物药代动力学研究数据;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]所述初步方案制定模块,用于利用导入的患者信息模块中的基本信息、药物基因检测模块中检测得到的患者抗脑卒中药物相关基因多态性信息、数据库模块中的治疗药物的临床使用信息,分析预估各种治疗方案对该患者个体的预期药效和不良反应风险,进一步判断候选药物与患者是否匹配,并确定患者的初步治疗方案;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]所述治疗信息更新模块,用于导入初步治疗方案和患者采用初步方案治疗后的最新复查结果;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]药物浓度监测模块,用于从患者样本中获得患者个体对抗脑卒中药物的体内暴露水平或代谢水平的信息;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]方案优化调整模块,用于利用导入的治疗信息更新模块中的数据、药物浓度监测模块监测得到的数据、数据库模块中的治疗药物的临床使用信息及临床药物药代动力学研究数据,根据患者复查结果以及实际药物暴露水平或代谢水平,综合分析评估初步治疗方案的治疗效果,井进一步对其优化调整,得到优化治疗方案。[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]第六目的在于提供一种指导脑卒中疾病个体化精准用药的方法,包括初步方案制定阶段和方案优化调整阶段;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]在初步方案制定阶段:[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]采集患者血浆样本,通过药物基因检测模块检测患者的抗脑卒中药物相关基因多态性信息;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]将药物基因检测模块中检测得到的数据、患者信息模块中的数据、数据库模块中的治疗药物的临床使用信息导入初步方案制定模块;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]初步方案制定模块根据导入的信息,分析预估各种治疗方案对该患者个体的预期药效和不良反应风险,进一步判断候选药物与患者是否匹配,并确定患者的初步治疗方案;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]在方案优化调整阶段:[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]将初步治疗方案和患者采用初步方案治疗后的最新复查结果导入治疗信息更新模块;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]采集患者血浆样本,通过药物浓度监测模块监测患者个体对抗脑卒中药物的体内暴露水平或代谢水平信息;[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]将治疗信息更新模块中的信息、药物浓度监测模块监测得到的信息、数据库模块中的治疗药物的临床使用信息及临床药物药代动力学研究数据,导入方案优化调整模块,方案优化调整模块根据导入的信息,根据患者复查结果以及实际药物暴露水平或代谢水平,综合分析评估初步治疗方案的治疗效果,并进一步对其优化调整,得到优化治疗方案。[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]进一步的,所述患者信息包括基本信息、疾病状态指标和肝肾功能指标。[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]进一步的,所述抗脑卒中药物相关基因多态性信息包括药物转运、代谢、药效和毒性作用相关的重要基因位点中的至少一种。[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left][font=宋体][color=black]进一步的,所述药物基因检测模块采用基质辅助激光解吸电离飞行时间质谱、[/color][/font][font='Times New Roman',serif][color=black]Sanger[/color][/font][font=宋体][color=black]测序和荧光定量[/color][/font][font='Times New Roman',serif][color=black]PCR[/color][/font][font=宋体][color=black]检测方法中的至少一种。[/color][/font][/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]进一步的,所述临床使用信息包含抗脑卒中药物临床指导原则、药物使用禁忌和药物之间相互作用。[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]进一步的,所述初步治疗方案还包括过往的参考治疗病例、注释该药物的用药禁忌和与其他药物的相互作用风险、下一步药物浓度监测实验设计方案。[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]进一步的,所述临床使用信息包含抗脑卒中药物临床指导原则、剂量调整方法、药物使用禁忌和药物之间相互作用。[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]进一步的,所述药物浓度监测模块的检测方法为液相色谱法、液相色谱质谱联用法、薄层色谱法和核磁定量法中的至少一种。[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]进一步的,所述优化调整后的治疗方案包括前期治疗方案存在的问题、优化的治疗方案及预期药效和不良反应风险。[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]本发明的有益效果在于:[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]本发明所述的脑卒中疾病临床用药指导系统,采用药物基因检测方法,可同时得到患者个体的预期药效和不良反应风险,进一步判断候选药物与忠者是否匹配,确定患者的初步治疗方案。根据前期的初步治疗方案和患者接受初步治疗后的复查结果,系统采用药物浓度监测方法,根据患者复查结果以及实际药物暴露水平或代谢水平,综合分析评估初步治疗方案的治疗效果,并进一步对其优化调整,得到优化治疗方案。[/align][font='微软雅黑',sans-serif][/font][align=left][/align][font=宋体][/font][align=left]采用所述的脑卒中疾病临床用药指导系统,可使个体化治疗方案的准确度更高,更加精确有效。系统提供的药物治疗方案制定覆盖了从初期诊断到后期治疗的全流程,可针对脑卒中的联合用药方案,对药物相互作用进行分析,选择合适的药物种类,降低潜在的安全风险。[/align]
临床用药实例分析——执业药师继续教育资料

 400-860-5168转4843
400-860-5168转4843
 留言咨询
留言咨询
 留言咨询
留言咨询
 400-860-5168转4645
留言咨询
400-860-5168转4645
留言咨询








 我要推广仪器
我要推广仪器
 下载APP
下载APP


