推荐厂家
暂无
暂无



古老病毒通过入侵重塑人类基因组译者:Docofsoul《每日科学》2010年9月13日报道 —— 新加坡基因组研究院(GIS, 隶属于新加坡科技研究局(A*STAR)的生物医学研究院)的科学家以及来自新加坡国立大学、新加坡南洋理工大学、杜克-新加坡大学医学研究院与普林斯顿大学的同事们最近发现:数百万年前“入侵”人类基因组的病毒已经改变了人类胚胎干细胞(ES细胞)中的基因开启与关闭方式。科学家已经发现数百万年前“入侵”人类基因组的病毒已经改变了人类胚胎干细胞(ES细胞)中的基因开启与关闭方式。(照片来源:iStockphoto/Martin McCarthy)这一研究为生理学与医学诺贝尔奖获得者芭芭拉•麦克林托克(Barbara McClintock)于上世纪五十年代提出的理论提供了明确的证据。芭芭拉•麦克林托克的理论推测:转座因子,即可移动的遗传物质(DNA)片段(比如说病毒序列),一旦插入基因组,就能成为影响基因调节的“控制因子”。本发现对于推进干细胞研究进程、增强干细胞研究为再生医学效劳的潜力都算得上是重要贡献。 由新加坡基因组研究院精英小组负责人吉拉姆•布尔克(Guillaume Bourque)博士率队领导了本研究。本研究的论文发表于2010年6月6日的《Nature Genetics》(《自然•遗传学》)。通过运用新的测序技术,科学家们研究了人类与小鼠胚胎干细胞(ES细胞)中三种调节蛋白质(OCT4、NANOG 与 CTCF) 的染色体组定位(基因组定位)。令人感兴趣的是,在科学家发现大量的相似点的同时,他们也发现了在人类中受到调控的基因方式与基因类型的许多不同点。尤其是,他们发现:数百万年前自行插入人类基因组的特定类型病毒已经戏剧性地改变了人类干细胞基因调控网络。德克萨斯州大学阿灵顿分校副教授Cedric Feschotte 博士说:“本研究是计算与实验双管齐下的代表作,提供了无可置疑的全新的证据:一些经常被斥责为纯粹垃圾DNA的转座因子,恰恰正是人类发育调控密码的关键成分。”在基因调控网络的研究中,人类模型系统与小鼠模型系统之间的比较研究有助于增进对干细胞分化成体内不同细胞类型的具体过程的理解。布尔克博士说:“这种理解在促使再生医学的百尺竿头更进一步地发展 —— 从而解决诸如帕金森病与白血病等问题方面是至关重要的。除了在本研究中利用基因调控网络中的小鼠胚胎干细胞的优势外, 深入研究必须更加直接地集中于人类干细胞。这是因为将某一种类上完成的研究成果转向对另一种类的研究上时必然会遇上的挑战。为了让干细胞方面的发现能够用于临床实践,在人类与(非人类的)灵长类干细胞两个方面还有更多的研究工作需要完成。” 加利福尼亚州立大学神经学Rudi Schmid 特聘教授、哲学博士雷蒙德•怀特(Raymond L. White)教授说:“本论文报告了令人非常激动的新发现,证实了一个全新的、迥然不同的基因表达的调控机制。通过将小鼠的基因组与人类基因组的直接比较,科学家能够显示:在两种种类之间,基因调控因子的结合点经常不在同一位置。这本身就足够令人惊讶的了,但是研究者作了进一步的探索,证实许多位点都嵌合在称之为‘转位’因子的一类DNA序列中,这是因为他们具有在基因组中移动到新的位置的能力。存在很多这样的相信是病毒基因组进化残余部分的因子,但我们所了解到的(信息中)还有着非常出人意外的情形:它们到达新的(基因组)位置时,还携带着调控因子结合位点。这些在调控方面的变化估计可能在携带它们的有机体上产生重大变化。确实,许多学者相信调控方面的变化处于物种形成的核心,可能在人从其祖先的进化历程中扮演了一个重要角色。本论文可能成为这一研究领域的里程碑式的论文。”美国能源部联合基因组研究所所长、劳伦期•伯克利国家实验室伯克利实验分室基因组学部主任埃迪•拉宾(Eddy Rubin)博士补充说:“这个运用了比较基因组学策略的研究在人类胚胎干细胞(ES细胞)中发现了重要的人类特异性属性。该论文所提供的信息意义重大,应该有助于推进再生医学领域的发展,相信会有不俗的积极表现。”参考文献:Galih Kunarso, Na-Yu Chia, Justin Jeyakani, Catalina Hwang, Xinyi Lu, Yun-Shen Chan, Huck-Hui Ng, Guillaume Bourque. Transposable elements have rewired the core regulatory network of human embryonic stem cells.Nature Genetics, 2010; 42 (7): 631 DOI: 10.1038/ng.600(《转位因子重新连接人类胚胎干细胞的核心调控网络》)
人类基因组单核苷酸多态性的研究进展与动态The research development of single nucleotide polymorphisms in human genome 摘要:第一张人类基因组序列草图已经公布,正式图预计也将于2003年4月完成。但序列图只基于少数个体,它反映了基因组稳定的一面,并未反映其变异或多态的一面,而正是这种多态性,即基因组序列的差异构成了不同个体与群体对疾病的易感性、对药物与环境因子不同反应的遗传学基础。人类基因组中存在广泛的多态性,最简单的多态形式是发生在基因组中的单个核苷酸的替代,即单核苷酸多态性(single nucleotide polymorphisms, SNPs)。SNP通常是一种二等位基因的(biallelic),即二态的遗传变异,在CG序列上出现最为频繁。在转录序列上的SNP称为cSNP。SNP的数量大、分布广。按照1%的频率估计,在人类基因组中每100~300个核苷酸就有一个SNP。因此,整个人类基因组(3.2 X 109bp)中至少有1,100万以上的SNPs,在任何已知或未知基因内和附近都可能找到数量不等的SNP 目前普遍认为,作为数量最多且易于批量检测的多态标记,SNP在连锁分析与基因定位,包括复杂疾病的基因定位、关联分析、个体和群体对环境致病因子与药物的易感性研究中将发挥愈来愈重要的作用。迄今,对多基因疾病候选基因的SNPs研究已积累了丰富的数据,基于这些SNPs的关联分析也正方兴未艾。本文阐述了SNP的特征、不同研究者对基于SNP进行关联分析的观点以及SNP的研究进展与动态。 关键词: SNP;遗传标记;关联研究 中图分类号:Q75 随着分子遗传学的进展,疾病遗传学研究从简单的单基因疾病转向于复杂的多基因疾病(如骨质疏松症、糖尿病、心血管疾病、精神性紊乱、各种肿瘤等)与药物基因组学的研究中。与前者相比,多基因性状或遗传病的形成,受许多对微效加性基因作用,即其中每种基因的作用相对较微弱。这些不同基因构成的遗传背景中,可能有易感性主基因(major gene)起着重要作用。它们同时还受环境因素的制约,彼此间相互作用错综复杂,所以任一基因的多态性对疾病发生仅起微弱的作用。鉴于此,需要在人类基因组中找到一种数目多、分布广泛且相对稳定的遗传标记,单核苷酸多态性(single nucleotide polymorphisms, SNPs)正是代表了这样一种标记,所以它成为继第一代限制性片段长度的多态性标记、第二代微卫星即简单的串联重复标记后,第三代基因遗传标记。 1. SNP作为遗传标记的优势 SNP自身的特性决定了它比其它两类多态标记更适合于对复杂性状与疾病的遗传解剖以及基于群体的基因识别等方面的研究。 (1)SNP数量多,分布广泛。据估计,人类基因组中每1000个核苷酸就有一个SNP,人类30亿碱基中共有300万以上的SNPs。SNP 遍布于整个人类基因组中,根据SNP在基因中的位置,可分为基因编码区SNPs(Coding-region SNPs,cSNPs)、基因周边SNPs(Perigenic SNPs,pSNPs)以及基因间SNPs(Intergenic SNPs,iSNPs)等三类。 (2)SNP适于快速、规模化筛查。组成DNA的碱基虽然有4种,但SNP一般只有两种碱基组成,所以它是一种二态的标记,即二等位基因(biallelic)。 由于SNP的二态性,非此即彼,在基因组筛选中SNPs往往只需+/-的分析,而不用分析片段的长度,这就利于发展自动化技术筛选或检测SNPs。主要的技术方法包括单链构象多态性(single strand conformation polymorphisms, SSCPs)法、异源双链分析(heteroduplex analysis, HA)、DNA直接测序分析、变异检测阵列(variant detector arrays, VDA)法以及基质辅助激光解吸附电离飞行时间(MALDI-TOF)质谱法等。 (3)SNP等位基因频率的容易估计。采用混和样本估算等位基因的频率是种高效快速的策略。该策略的原理是:首先选择参考样本制作标准曲线,然后将待测的混和样本与标准曲线进行比较,根据所得信号的比例确定混和样本中各种等位基因的频率。 (4)易于基因分型。SNPs 的二态性,也有利于对其进行基因分型。对SNP进行基因分型包括三方面的内容:(1)鉴别基因型所采用的化学反应,常用的技术手段包括:DNA分子杂交、引物延伸、等位基因特异的寡核苷酸连接反应、侧翼探针切割反应以及基于这些方法的变通技术;(2)完成这些化学反应所采用的模式,包括液相反应、固相支持物上进行的反应以及二者皆有的反应。(3)化学反应结束后,需要应用生物技术系统检测反应结果。目前许多生物技术公司发展出高通量检测SNP的技术系统,如荧光微阵列系统(Affymetrix)、荧光磁珠技术(Luminex,Illumina, Q-dot)、自动酶联免疫(ELISA)试验(Orchid Biocomputer)、焦磷酸的荧光检测(Pyrosequencing)、荧光共振能量转移(FRET)(Third Wave Technologies)以及质谱检测技术(Rapigene, Sequenom)。 2. 基于SNP的关联研究 如果某一因素可增加某种疾病的发生风险,即与正常对照人群相比,该因素在疾病人群中的频率较高,此时就认为该因素与疾病相关联。如非遗传因素吸烟与肺癌相关;在遗传因素中,如APOE4与Alzheimer`s相关。对疾病进行关联分析需要在年龄与种族相匹配的患者和对照人群中确定待测因素(环境的或遗传的)的频率分布,患者和对照人群的选择是否恰当直接影响结果的可靠性。对常见的由高频率、低风险等位基因导致的疾病,采用致病等位基因的关联分析比连锁分析更有效。 应用SNP进行关联研究,首先需明确多少SNPs才可满足在全基因组范围内的分析。Kruglyak应用计算机模拟法预测人类基因组中超过3Kb就不存在连锁不平衡,据此推出完成全基因组扫描将需要500,000个SNPs。而Collins等收集通过家系研究得到的常染色体单倍型的信息发现,在染色体上相距0.2cM到0.4cM(约200-400kb)之间的标记仍存在连锁不平衡,如按每100kb需要一个SNP计算,那么完成全基因组扫描仅需约30,000个SNPs,平均每3-4个基因用一个SNP就可识别出整个基因组内任何位置上的具表型活性的变异。最近发现SNP与SNP之间的连锁不平衡甚至可延伸到更远的区域(0.35cM-0.45cM),那么进行基因组扫描需要的SNP数量就更少。导致上述估算SNP 数量差异的主要原因是Kruglyak进行模拟计算时,假设现在的人群在5000年前起源于共同的祖先,且人群规模的有效大小保持在10,000左右,然后经过连续的指数扩增,直至达到现在的50亿左右。Collins认为这种假设是不现实的,在人类发展的历史过程中,人群数目的增长是迂回曲折的,经历扩张与萎缩的周期性变化。 Weiss等认为Collins及其同事的结果可能低估了问题的复杂性。因为他们的结果或是基于小样本资料推断出来的,就会使连锁不平衡(LD)程度的估算偏高;或是从理论上预测LD的水平,而忽略了基因组中大量的随机变异。如大多数位点的信息是来源于小样本中测序得到的资料,据此得到的单倍型结构不可靠。目前的研究集中于基因组中LD相对广泛存在的区域,在此区域内,基因相对容易作图。如基于这些经验来进行基因组其它区域的LD分析,就可能发生偏离。如两个相距较远的SNPs 之间具有强的LD性质,就认为它们之间的SNPs及该SNP侧翼的SNPs也存在强烈的LD,这种假设仅适合于其中一些多态位点,但它并不是通则。当然,在一些罕见人群中,如Saami,在较长的区域内广泛存在大量的LD,但对Fihland人群,则在较长区域内几乎不存在LD,对全球整个复杂人群而言,LD肯定变得更复杂一些。 Gray等认为随着人类基因组测序计划的进展,人类基因组的结构逐渐被阐明,因此就可在那些富含基因的区域选择SNP进行全基因组扫描,这样所需的SNP数量还会减少。Halushka等根据他们对75个基因检测的实验结果推测,SNPs在单个基因或整个基因组中的分布是不均匀的,在非转录序列中要多于转录序列,而且在转录区也是非同义突变的频率比其它方式突变的频率低得多。Templeton 等对LPL基因突变与重组热点的研究结果提示,SNP集中分布于基因组的CG二核苷酸处或单核苷酸重复区或αDNA聚合酶的识别位点(TGGA)处。将人类基因组不同区域物理图谱与遗传图谱的进行比较,发现遗传距离和物理距离的比值有很大的差异,提示基因组不同区域的重组水平存在差异。如Dunham等将22号染色体STR的物理位置与遗传位置进行了对比,发现该染色体的重组率差异很大,提示存在重组热点。根据基因组内不同区域重组频率的高低可进一步选择SNP的数量,重组热点需要的标记数量就多,相反就少。这种设计也可能会进一步减少基因组扫描所需的SNP标记。 使用SNP进行关联分析面临的另一个问题是如何选择SNP。如果对每一个SNP都进行独立研究,那么对几百万SNPs 的研究就会导致成千上万次的假关联,结果就掩盖真实的关联性,所以,进行关联分析前,一定要对所研究的SNP进行选
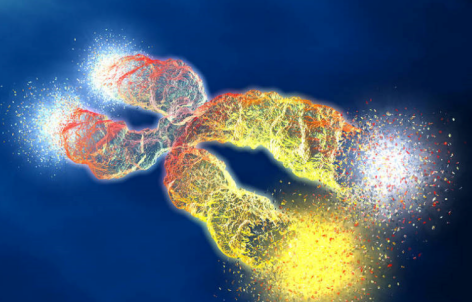
北京时间12月21日消息,美国《科学》杂志12月21日公布了2007年度科学突破,“科学家发现人类基因组差异”荣登榜首,成为2007年度最大的科学突破。以下是《科学》杂志年度十大科学突破名单:[B][size=4]1.揭开人类基因组个体差异之谜[/size][/B][img]http://ng1.17img.cn/bbsfiles/images/2007/12/200712230429_74133_1622715_3.jpg[/img]揭开人类基因组个体差异之谜在更为先进的DNA排序技术和基因组个体差异评估技术的帮助下,研究人员正在逐步揭开人与人之间差异的谜底。7年前科学家成功破译人类基因组,为首次揭示人类完整的基因构成奠定了基础。到了2007年,研究人员逐步意识到人与人之间基因组差异到底有多大,以及这种差异对破译复杂疾病和个人性格的重要性。差不多一年前,科学家又获得重要发现,加深了对人类和灵长类动物之间基因差异的认识,对最终导致人类出现的进化过程的基因变化有了深入了解。如今,科学家的研究重点已从寻找DNA对群体影响的答案转向寻找DNA对个体影响的答案。曾用于寻找数十万基因差异的高科技现在正以一种前所未有的方式,将特定差异与疾病联系起来。科学家通过评估染色体在我们人类DNA突增和缺失,结果发现这些变化比他们预料的更为普遍,与人类基因组的运转密切相关。通过研究决定头发、皮肤颜色的基因以及“语言”基因差异,我们已经对人类与穴居人的不同和相同之处有了深入了解。随着个体基因差异谜团的逐步揭开,我们势必会在这个领域取得巨大飞跃。

 400-860-5168转4819
400-860-5168转4819
 留言咨询
留言咨询
 400-801-8117
留言咨询
400-801-8117
留言咨询
 400-807-5250
留言咨询
400-807-5250
留言咨询









 我要推广仪器
我要推广仪器
 下载APP
下载APP


