

方案详情文
智能文字提取功能测试中
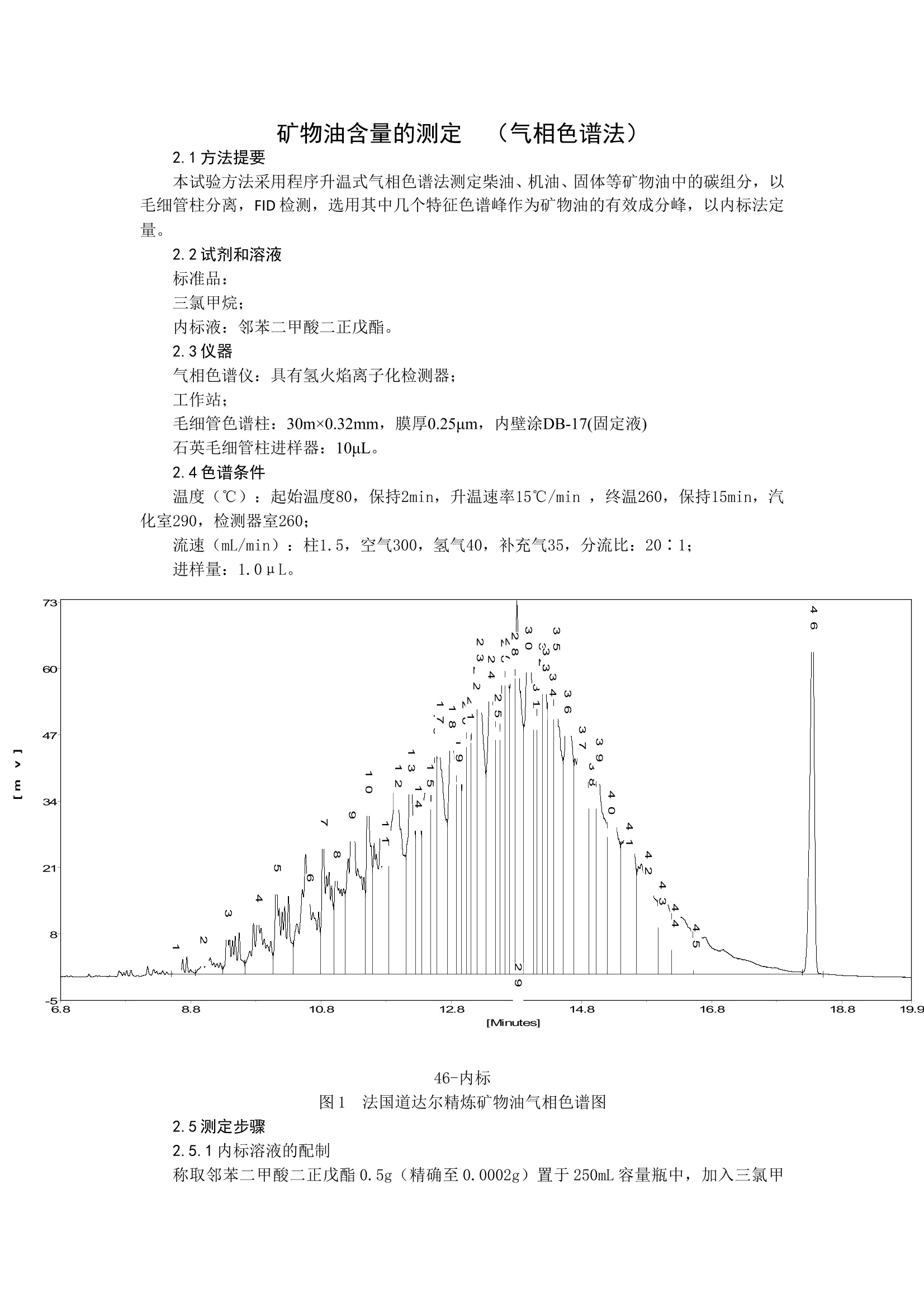
矿物油含量的测定 (气相色谱法) 2.1方法提要 本试验方法采用程序升温式气相色谱法测定柴油、机油、固体等矿物油中的碳组分,以毛细管柱分离,FID检测,选用其中几个特征色谱峰作为矿物油的有效成分峰,以内标法定量。 2.2试剂和溶液 标准品: 三氯甲烷; 内标液:邻苯二甲酸二正戊酯。 2.3仪器 气相色谱仪:具有氢火焰离子化检测器; 工作站; 毛细管色谱柱:30m×0.32mm,膜厚0.25μm,内壁涂DB-17(固定液) 石英毛细管柱进样器:10μL。 2.4色谱条件 温度(℃):起始温度80,保持2min,升温速率15℃/min ,终温260,保持15min,汽化室290,检测器室260; 流速(mL/min):柱1.5,空气300,氢气40,补充气35,分流比:20∶1; 进样量:1.0μL。 46-内标 图1 法国道达尔精炼矿物油气相色谱图 2.5测定步骤 2.5.1内标溶液的配制 称取邻苯二甲酸二正戊酯0.5g(精确至0.0002g)置于250mL容量瓶中,加入三氯甲烷使其溶解,定容摇匀,备用。 2.5.2标样溶液的配制 称取矿物油标准品0.2g(精确至0.0002g)置于25mL具塞小锥形瓶中,准确吸取25mL内标溶液使其溶解,摇匀,备用。 2.5.3样品溶液的配制 称取含矿物油标准品约0.2g(精确至0.0002g)置于25mL具塞小锥形瓶中,准确吸取25mL内标溶液使其溶解,摇匀,备用。 2.5.4测定 在上述色谱操作条件下,待仪器稳定后,先进数针标样溶液直至相邻两针相对响应值变化不大于1.5%时,按标准溶液、样品溶液、样品溶液、标准溶液顺序进样。 2.6计算 将测得的两针试样溶液以及前后两针标样中柴油(或其它矿物油)峰面积(几种特征峰面积之和)与内标物峰面积比分别进行平均。试样中柴油(或其它矿物油)质量分数X1(%),按式(1)计算: r2M1p X1(%)= ………………………………………………(1) r1M2 式中: X1 试样中柴油(或其它矿物油)的含量,%。 r1 两针样品溶液中柴油(或其它矿物油)峰面积(几种特征色谱峰面积之和)与内标物峰面积比的平均值; M2 柴油(或其它矿物油)标准品的质量,g; P 标准品中柴油(或其它矿物油)的含量,%。 r1 两针标准溶液中柴油(或其它矿物油)峰面积(几种特征色谱峰面积之和)与内标物峰面积比的平均值; M1 样品质量,g; 计算结果应表示至两位小数。 2.7允许差 两次平行测定结果之差不大于a%(a应根据产品中矿物油的质量分数而定)。 2.8 方法讨论 2.8.1 本方法仅适用于农药产品中柴油、石蜡、机油等矿物油含量的测试。 2.8.2 对不同矿物油的定量测定方法中仪器色谱条件和内标物的选择是典型的,操作者可根据仪器不同特点对操作参数和内标物的类型做适当调整,以期获得最佳分离效果。 2.8.3 机油的气相色谱图中本底吸收较高,对含量的准确定量影响较大;韩国SK矿物油气相色谱图中本底吸收太高,定量时误差较大。同时对于本底吸收较高的其他矿物油产品,控制项目指标中碳数范围、碳数差、平均碳数的测定会带来一定误差。部分产品碳数范围测定时,经查沸点-保留时间曲线图与GS-MS定性测得的数据存在一定差距。 3. 相对正构烷烃碳数范围差及平均碳数 该项质量控制项目指标测定方法主要参考 ASTM2887 或 SH/T0558-93 标准,在现有仪器设备某些功能的条件下,操作步骤进行了简化,更便于试验操作。 3.1 方法提要 本试验方法采用程序升温式气相色谱法测定正构烷烃,取得保留时间和沸点数据,绘制沸点-相对保留时间曲线图;同时测定矿物油的相对保留时间,通过查阅曲线图,得到待测矿物油的相对碳数范围差及平均碳数;以FID为检测器。 3.2 试剂和溶液 样品:C12、C16、C20、C22、C24,矿物油; 三氯甲烷; 3.3 仪器 气相色谱仪:具有氢火焰离子化检测器; 工作站; 毛细管色谱柱:30m×0.32mm,膜厚0.25μm,内壁涂DB-17(固定液) 石英毛细管柱进样器:10μL。 色谱条件 温度(℃):起始温度80,保持2min,升温速率15℃/min ,终温260,保持15min,汽化室290,检测器室260; 流速(mL/min):柱1.5,空气300,氢气40,补充气35,分流比:20∶1; 进样量:1.0μL。 测定步骤 标样溶液的配制 称取C12、C16、C20、C22、C24等正构烷烃约0.05g(精确至0.0001g)置于25mL容量瓶中,用三氯甲烷溶解,摇匀,备用。 3.5.2样品溶液的配制 称取矿物油样品0.05g(精确至0.0001g)置于25mL容量瓶中,用三氯甲烷溶解,摇匀,备用。 3.5.3测定 在上述色谱操作条件下,待仪器稳定后,先进数针标样溶液直至相邻两针响应值变化小于1.5%时,按C12、C16、C20、C22、C24等正构烷烃顺序,分别进两针,再进样品溶液两针。 下图为典型正构烷烃气相色谱图 1-C12 2-C16 3-C20 4-C22 5-C25 图2 正构烷烃色谱图 3.5.4绘制工作曲线 对上述色谱操作条件下取得的正构烷烃标样溶液的两针相对保留时间分别进行平均,并计算该相对保留时间处所对应的沸点,取得各点的(t,℃)数据,绘制沸点(℃)-保留时间(min)曲线图,见图3 图4 正构烷烃的沸点-保留时间曲线图 3.5.5计算并查阅结果 取矿物油样品的面积归一结果,通过分别计算10%和90%面积百分比所对应的相对保留时间,查阅上述沸点-保留时间曲线图,查得试样在10%溜出温度与90%溜出温度所相对的正构烷烃碳数,即为试样的碳数范围;计算相对的正构烷烃碳数之差即为碳数差。同时计算试样在50%溜出温度相对的平均正构烷烃碳数即为平均碳数。 相对正构烷烃碳数是根据在某个馏分相当的正构烷烃来区分的,并不是非常严格意义上的化学组分,因为大于C16的正构烷烃是固体石蜡,而他们并不存在于矿物油中。该项指标是指在所规定的气相色谱条件下,试样90%溜出温度与10%溜出温度所相对的正构烷烃碳数之差。控制该项指标主要是为了对产品中组分的沸程范围进行评价,以降低药害的风险。在发达国家,通常的矿物油的相对正构烷烃碳数差应当≤7.考虑分析误差,将该项指标定为≤8. 该方法即可用于测定矿物油单制剂产品,也可以用于矿物油与其他农药混配的产品。采用工作曲线法查得的碳数范围可能存在一定误差,工作曲线的绘制一定要用连续的正构烷烃测定,以减少测定误差。 4.非磺化物含量 参照ASTM D483-92 标准和石化行业 SYB2115-59 标准(该标准已更新为SH/T 0117-92),对试验操作步骤进行了简化,更便于实际操作。 4.1 仪器与试剂 磺化器:带磨口塞的特制量筒120mL(量筒90mL以下部分,直径为5cm,90mL以上部分具刻度,直径为1cm); 量筒:100mL; 浓硫酸:分析纯。 4.2 测定步骤 用量筒量取浓硫酸90mL注入干燥磺化器中,再准确加入15mL试样,剧烈震摇5min,然后将磺化器垂直静置4~5小时,待磺化器中液体分为两层,准确记录上层矿物油的体积。 4.3 计算 非硫酸化残余物的含量X2(%)按(2)式计算: V1 X2=————×100··················(2) V0 式中: X2 ———矿物油中非硫酸化残余物的体积百分含量,%; V1———上层矿物油的体积, V; V0———试样的体积, V; 4.4 允许差 两次平行测定结果之差应不大于0.5%。 矿物油含量的测定 (气相色谱法)2.1方法提要本试验方法采用程序升温式气相色谱法测定柴油、机油、固体等矿物油中的碳组分,以毛细管柱分离,FID检测,选用其中几个特征色谱峰作为矿物油的有效成分峰,以内标法定量。2.2试剂和溶液标准品:三氯甲烷;内标液:邻苯二甲酸二正戊酯。2.3仪器气相色谱仪:具有氢火焰离子化检测器;工作站;毛细管色谱柱:30m×0.32mm,膜厚0.25μm,内壁涂DB-17(固定液)石英毛细管柱进样器:10μL。2.4色谱条件温度(℃):起始温度80,保持2min,升温速率15℃/min ,终温260,保持15min,汽化室290,检测器室260;流速(mL/min):柱1.5,空气300,氢气40,补充气35,分流比:20∶1;进样量:1.0μL。 46-内标图1 法国道达尔精炼矿物油气相色谱图2.5测定步骤2.5.1内标溶液的配制称取邻苯二甲酸二正戊酯0.5g(精确至0.0002g)置于250mL容量瓶中,加入三氯甲烷使其溶解,定容摇匀,备用。2.5.2标样溶液的配制称取矿物油标准品0.2g(精确至0.0002g)置于25mL具塞小锥形瓶中,准确吸取25mL内标溶液使其溶解,摇匀,备用。2.5.3样品溶液的配制称取含矿物油标准品约0.2g(精确至0.0002g)置于25mL具塞小锥形瓶中,准确吸取25mL内标溶液使其溶解,摇匀,备用。2.5.4测定在上述色谱操作条件下,待仪器稳定后,先进数针标样溶液直至相邻两针相对响应值变化不大于1.5%时,按标准溶液、样品溶液、样品溶液、标准溶液顺序进样。2.6计算将测得的两针试样溶液以及前后两针标样中柴油(或其它矿物油)峰面积(几种特征峰面积之和)与内标物峰面积比分别进行平均。试样中柴油(或其它矿物油)质量分数X1(%),按式(1)计算: r2M1p X1(%)= ------ ………………………………………………(1) r1M2式中:X1 试样中柴油(或其它矿物油)的含量,%。r1 两针样品溶液中柴油(或其它矿物油)峰面积(几种特征色谱峰面积之和)与内标物峰面积比的平均值;M2 柴油(或其它矿物油)标准品的质量,g;P 标准品中柴油(或其它矿物油)的含量,%。r1 两针标准溶液中柴油(或其它矿物油)峰面积(几种特征色谱峰面积之和)与内标物峰面积比的平均值;M1 样品质量,g;计算结果应表示至两位小数。2.7允许差两次平行测定结果之差不大于a%(a应根据产品中矿物油的质量分数而定)。 2.8 方法讨论2.8.1 本方法仅适用于农药产品中柴油、石蜡、机油等矿物油含量的测试。2.8.2 对不同矿物油的定量测定方法中仪器色谱条件和内标物的选择是典型的,操作者可根据仪器不同特点对操作参数和内标物的类型做适当调整,以期获得最佳分离效果。2.8.3 机油的气相色谱图中本底吸收较高,对含量的准确定量影响较大;韩国SK矿物油气相色谱图中本底吸收太高,定量时误差较大。同时对于本底吸收较高的其他矿物油产品,控制项目指标中碳数范围、碳数差、平均碳数的测定会带来一定误差。部分产品碳数范围测定时,经查沸点-保留时间曲线图与GS-MS定性测得的数据存在一定差距。 3. 相对正构烷烃碳数范围差及平均碳数该项质量控制项目指标测定方法主要参考 ASTM2887 或 SH/T0558-93 标准,在现有仪器设备某些功能的条件下,操作步骤进行了简化,更便于试验操作。3.1 方法提要 本试验方法采用程序升温式气相色谱法测定正构烷烃,取得保留时间和沸点数据,绘制沸点-相对保留时间曲线图;同时测定矿物油的相对保留时间,通过查阅曲线图,得到待测矿物油的相对碳数范围差及平均碳数;以FID为检测器。3.2 试剂和溶液 样品:C12、C16、C20、C22、C24,矿物油; 三氯甲烷; 3.3 仪器 气相色谱仪:具有氢火焰离子化检测器;工作站;毛细管色谱柱:30m×0.32mm,膜厚0.25μm,内壁涂DB-17(固定液)石英毛细管柱进样器:10μL。3.4 色谱条件温度(℃):起始温度80,保持2min,升温速率15℃/min ,终温260,保持15min,汽化室290,检测器室260;流速(mL/min):柱1.5,空气300,氢气40,补充气35,分流比:20∶1;进样量:1.0μL。3.5 测定步骤3.5.1 标样溶液的配制称取C12、C16、C20、C22、C24等正构烷烃约0.05g(精确至0.0001g)置于25mL容量瓶中,用三氯甲烷溶解,摇匀,备用。3.5.2样品溶液的配制称取矿物油样品0.05g(精确至0.0001g)置于25mL容量瓶中,用三氯甲烷溶解,摇匀,备用。3.5.3测定在上述色谱操作条件下,待仪器稳定后,先进数针标样溶液直至相邻两针响应值变化小于1.5%时,按C12、C16、C20、C22、C24等正构烷烃顺序,分别进两针,再进样品溶液两针。下图为典型正构烷烃气相色谱图 1-C12 2-C16 3-C20 4-C22 5-C25图2 正构烷烃色谱图3.5.4绘制工作曲线对上述色谱操作条件下取得的正构烷烃标样溶液的两针相对保留时间分别进行平均,并计算该相对保留时间处所对应的沸点,取得各点的(t,℃)数据,绘制沸点(℃)-保留时间(min)曲线图,见图3 图4 正构烷烃的沸点-保留时间曲线图3.5.5计算并查阅结果取矿物油样品的面积归一结果,通过分别计算10%和90%面积百分比所对应的相对保留时间,查阅上述沸点-保留时间曲线图,查得试样在10%溜出温度与90%溜出温度所相对的正构烷烃碳数,即为试样的碳数范围;计算相对的正构烷烃碳数之差即为碳数差。同时计算试样在50%溜出温度相对的平均正构烷烃碳数即为平均碳数。 相对正构烷烃碳数是根据在某个馏分相当的正构烷烃来区分的,并不是非常严格意义上的化学组分,因为大于C16的正构烷烃是固体石蜡,而他们并不存在于矿物油中。该项指标是指在所规定的气相色谱条件下,试样90%溜出温度与10%溜出温度所相对的正构烷烃碳数之差。控制该项指标主要是为了对产品中组分的沸程范围进行评价,以降低药害的风险。在发达国家,通常的矿物油的相对正构烷烃碳数差应当≤7.考虑分析误差,将该项指标定为≤8.该方法即可用于测定矿物油单制剂产品,也可以用于矿物油与其他农药混配的产品。采用工作曲线法查得的碳数范围可能存在一定误差,工作曲线的绘制一定要用连续的正构烷烃测定,以减少测定误差。 4.非磺化物含量参照ASTM D483-92 标准和石化行业 SYB2115-59 标准(该标准已更新为SH/T 0117-92),对试验操作步骤进行了简化,更便于实际操作。4.1 仪器与试剂 磺化器:带磨口塞的特制量筒120mL(量筒90mL以下部分,直径为5cm,90mL以上部分具刻度,直径为1cm);量筒:100mL;浓硫酸:分析纯。4.2 测定步骤用量筒量取浓硫酸90mL注入干燥磺化器中,再准确加入15mL试样,剧烈震摇5min,然后将磺化器垂直静置4~5小时,待磺化器中液体分为两层,准确记录上层矿物油的体积。4.3 计算非硫酸化残余物的含量X2(%)按(2)式计算: V1 X2=————×100··················(2) V0式中:X2 ———矿物油中非硫酸化残余物的体积百分含量,%;V1———上层矿物油的体积, V;V0———试样的体积, V;4.4 允许差两次平行测定结果之差应不大于0.5%。
关闭-
1/5
-
2/5

还剩3页未读,是否继续阅读?
继续免费阅读全文
北京希望世纪科技有限公司为您提供《含油污水中矿物油检测方案(测油仪)》,该方案主要用于废水中有机污染物检测,参考标准《暂无》,《含油污水中矿物油检测方案(测油仪)》用到的仪器有红外测油仪、日本HORIBA OCMA测油仪 H-997萃取液。
我要纠错



相关方案
 咨询
咨询




