



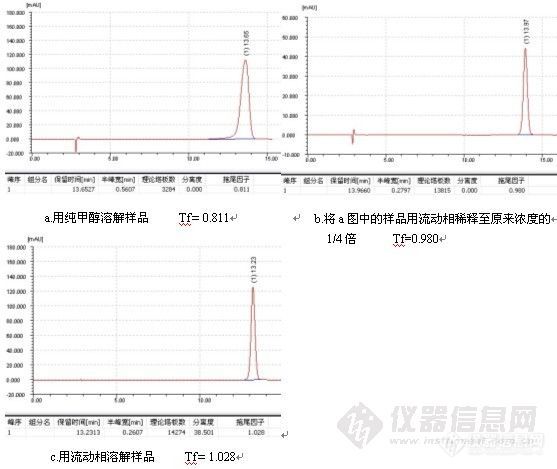
附件:
zxhcnf
第1楼2009/10/30
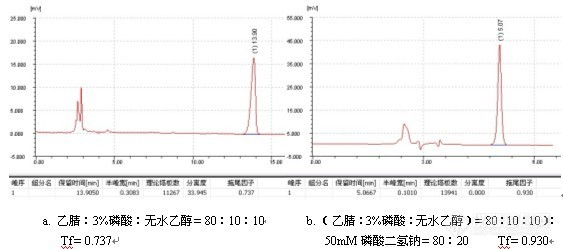
3)增加流动相中缓冲盐的浓度
增加缓冲盐浓度可以增大流动相中的离子强度,减少因静电的作用(有可能存在于样品分子之间、也有可能存在于样品分子与填料表面之间)引起的前拖。例子如下:
注射用苦参碱的测定
色谱柱:Ultimate XB-NH2-2, 5um, 4.6×250mm;
柱 温:室温24度; 流速:1.0ml/min;
检测波长:220nm; 进样量:20ul;
4)流动相中加入适量的四氢呋喃
往流动相中加入少量的四氢呋喃有时可以改善峰形、增大分离度,很多色谱工作者都知道和使用,但其机理似乎少人提及。通常所加入的量在5%以内即可,需要的时候可以加入更大的量。例子如下:
阿莫西林胶囊的测定
色谱柱:Ultimate AQ-C18,5um,4.6×250mm;
检测波长:254nm; 温度:室温28度;
流 速:1.0ml/min; 进样量:20ul;

5)升高柱温
升温有助于增加流动相传质速率,减少因静电作用引起的前拖,但温度不宜太高,温度太高容易损伤色谱柱,特别是含有离子对试剂的时候,最好不要超过40度。

































