小木
第2楼2009/12/06
2.2 色谱质谱方法的建立
2.2.1 质谱方法的建立
通常,发展质谱方法可采用直接注射法、进样泵法和液相色谱导入法。由于我们在试验中应用的是3200 Q-TRAP这款仪器,它自带进样泵,因此我们采用进样泵法。
首先,根据化合物A的性质,我们选用DMSO作为溶解样品的溶剂。为什么选择这个溶剂呢?因为它难挥发,这样就保证了样品浓度的一致性;因为溶解性好,基本能溶解所有的非盐类化合物。接下来将化合物A和内标B制备成浓度均为300ng/mL的溶解,用于质谱方法的建立。
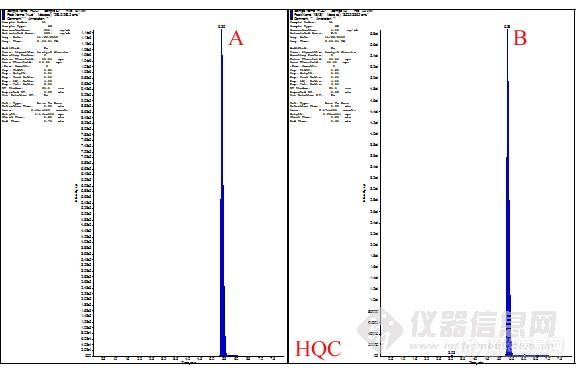
第一步,对化合物A进行Q1扫描。在这步操作中,主要优化DP电压,确定A的母离子,原则就是尽量选择分子离子峰——稳定,结果重现性好。
第二步,确定子离子和合适的碰撞能。确定了母离子后,进行子离子扫描。在选择子离子的过程中,可能会出现两个、三个甚至更多的子离子,丰度较大。遇到这种情况,不要着急确定哪个子离子更为合适,可以先留着,在进行infusion的时候,将这几个离子对都输入进去,经过仪器的自动优化,看看哪个离子对的响应强度更高,更为稳定。
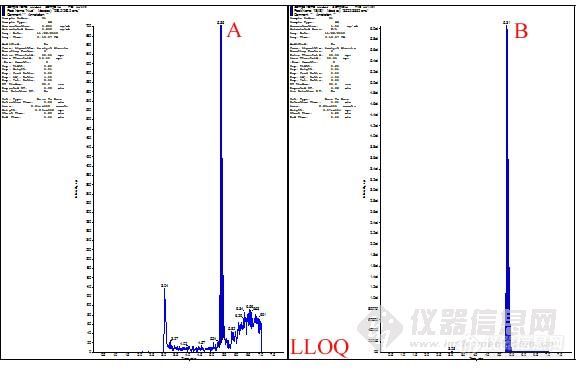
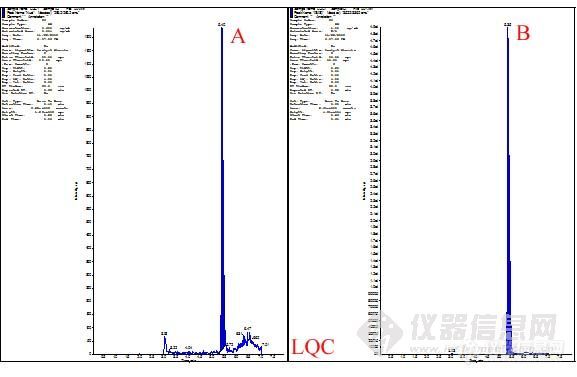
第三步,infusion和FIA。由前面两步,我们可以初步确定用于定量检测和定性检测的离子对,然后进行infusion操作。这步操作主要用于确定基本的质谱参数,如DP、CXP、CEP、EP等。上述参数确定后,保存此方法,连上HPLC,设定HPLC洗脱程序,进行FIA。此操作主要是系统的优化源参数和化合物的参数。优化完毕后,系统会自动的保存一个方法。至此,化合物A的质谱方法建立完毕。
内标B的优化方法同A。
质谱方法如下: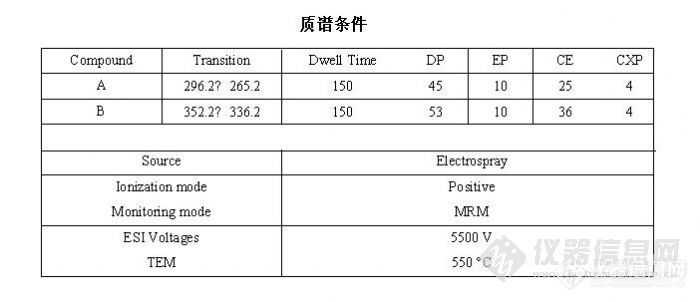
小木
第3楼2009/12/06
2.2.2 色谱方法的建立
第一步,添加剂的选择
在优化色谱条件时,首先考虑选择何种添加剂。由于化合物A呈弱碱性,而且我们采用正离子作为检测模式,所以为了保证化合物既在色谱柱上保留,又能够充分的进行离子化,选择0.1%的甲酸作为水相和有机相的添加剂。
第二步,色谱柱的选择
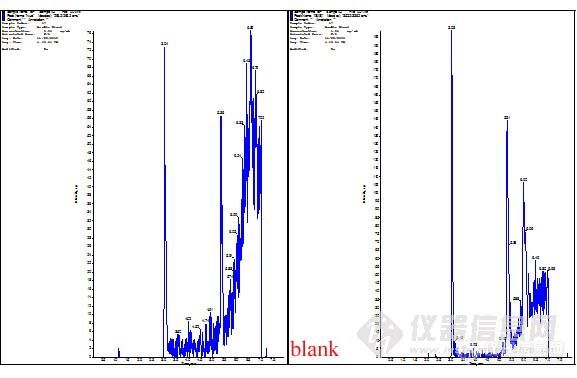
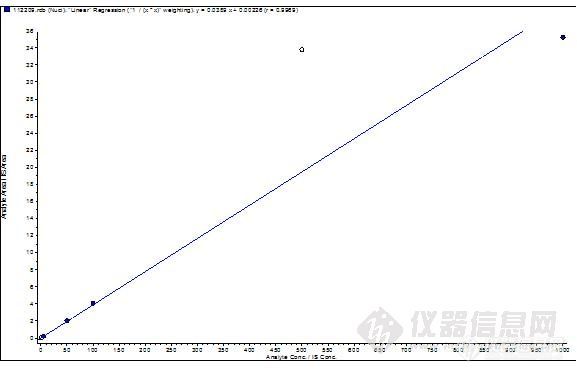
在确定添加剂后,我们对色谱柱进行了考察。手头主要有两款,一是C18 (5μm, 2.1*150mm),另一款是Ultimate LP—C18 (5μm, 3.0*100mm)。在使用C18色谱柱时,流速为0.4mL/min、甲醇-水(0.1%甲酸)比例为70:30的条件下,系统压力大约在150bar左右。产生这么高的压力,可能原因有两个——一是柱子的内径为2.1um的;二是这款柱子相对于质谱常用柱而言,长度比较长,从而导致柱压较高。我们尝试了等度和梯度洗脱两种方式,但色谱峰的峰形均不理想。
色谱图如下:
后来我们看到月旭公司正在仪器信息网搞色谱柱免费申请试用活动,遂联系月旭公司相关负责人。他们根据我试验的具体情况,向我推荐了Ultimate LP—C18 (5μm, 3.0*100mm)这款色谱柱。接上色谱柱,按照说明书进行了柱子活化。
第三步,洗脱方式的选择
然后考察了等度和梯度洗脱对色谱峰的影响。在等度洗脱条件下,甲醇-水在50:50时,化合物和内标在1min内出峰,这在进行生物样品分析时很难祛除内源性物质的干扰,也就无法避免基质效应对试验的影响。我们尝试增加水相的比例,但水相高的情况下化合物进入离子源后离子化效率降低,从而导致灵敏度降低。
等度洗脱色谱图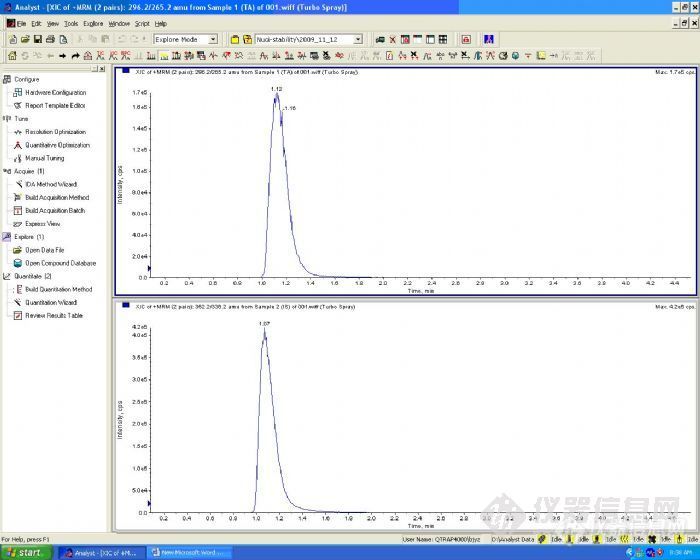
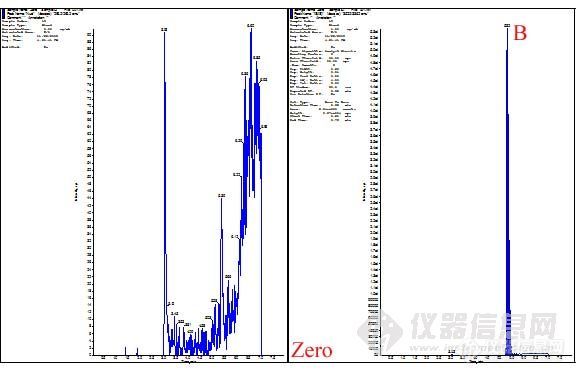
接下来我们考察了梯度洗脱,在初始流动相-水相为90%时,化合物A和内标B在6min左右出峰,总分析时长是10min。对于生物样品分析尤其是液质联用的高通量分析,分析时长太长。我们继续对这个洗脱条件进行优化,最后将A和B的出峰时间调节到5min左右。
梯度洗脱色谱图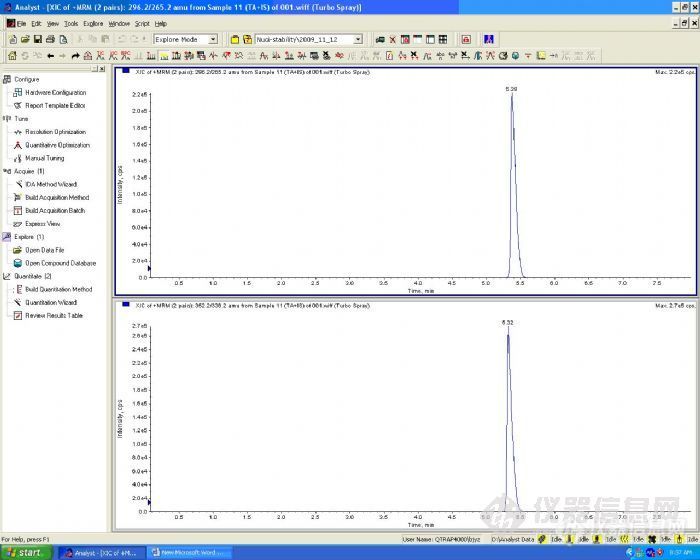
色谱方法如下: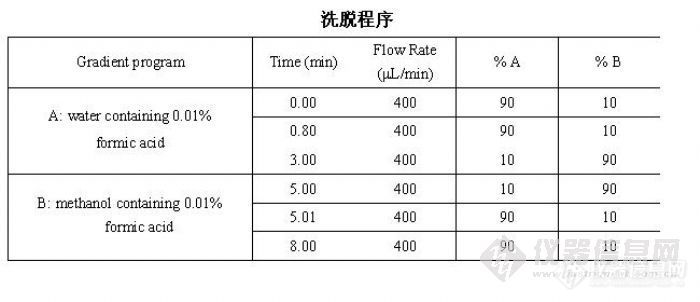
小木
第4楼2009/12/06
2.3 样品预处理方法的建立
常用的样品预处理方法主要有沉淀蛋白法、液液萃取法和固相萃取法。由于第三种由于固相萃取小柱价格昂贵,老板不给购买~~~,这里主要讨论前面两种方法。
首先,我们考察了沉淀蛋白法。沉淀试剂主要是甲醇和乙腈,考察了2倍量、三倍量对提取回收率的影响。结果显示,经这两种沉淀试剂处理后进样,乙腈三倍量提取后的化合物A和内标B的峰高高于其他处理方法的。但是所有的色谱图背景噪音(基线)较大,而且内源性物质干扰很明显。同时,鉴于化合物的化学性质,也考察了碱化试剂对提取回收率的影响。结果显示,碱化试剂对其响应影响不大。
在上述情况下,我们考察了液液提取法。提取试剂主要有甲基叔丁基醚,正己烷,二氯甲烷,乙酸乙酯以及正己烷-二氯甲烷3:1等。结果显示,乙酸乙酯的提取回收率最高,这可有相似相容原理来解释。使用乙酸乙酯唯一的缺点就是——氮气吹干浓缩时,浓缩速率慢,成了整个试验的限速步骤。
氮吹仪装置图