化学药检测
小卢
第1楼2010/08/12
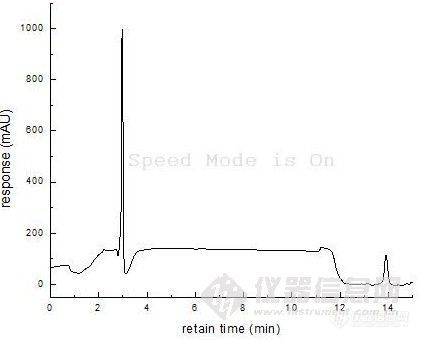
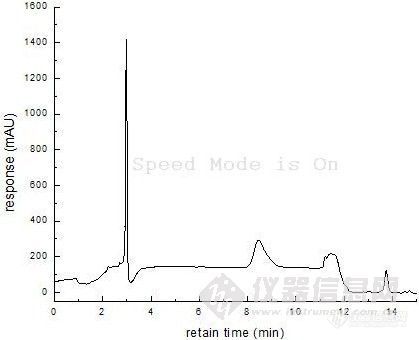
基线走的很不稳啊,是13.8min左右的峰吗?你计算时的稀释倍数搞清楚了没?
lwjxzj
第2楼2010/08/12
以下是做标曲的原始数据5 721.27.5 861.310 945.412.5 1011.815 1090.725 1373.9HPLC整个过程采用梯度洗脱,条件是:time(min) 乙腈(%) 0.05%三氟乙酸(%)0 10 908 18 828.1 100 012 100 015 10 90210nm检测
第3楼2010/08/12
是3min的那个很高的峰请问如何让出峰时间延迟呢?请问什么叫“样品好像在死体积附近”,我应该怎么改进方法呢?“改善分离条件,如使用离子对是试剂 ...”请问什么是“离子对是试剂”呢?标准曲线的横坐标是表示A物质的质量,我的计算过程是(1280.1-609.64)/31.286=21ug 刚才,把一楼的结果写错了,已经改过来了,呵呵不过还是21ug〉5ug,请大家再帮我想想这是为什么呢?
xiaohe147
第4楼2010/08/17
1.感觉好像是浓度配制错了。如果对照品和样品一起配错了,标准曲线的横坐标就是错的,结果肯定有问题。2.A在柱子上没什么保留,在死体积出峰。可以尝试将梯度起始时间的水相调整为0,从100%的水相开始变化,A峰的保留时间应该会增加。
jm0311
第5楼2011/01/12
我觉得问题出在你的目标峰出在死时间附近的,线性是很不好的,R2=0.98就说明这个问题,赞成4楼的意见,调流动相,如果可以的话换根长点的柱子。降低柱温,还有的话进针溶剂扣下空白,再做曲线看看。还是推荐采用峰面积作曲线。
lunanjituan
第6楼2011/01/20
一、你的标准品含量100%的吗?二、按照你的峰高看是在结果20μg左右的,这个应该不会错三、用外标法以峰面积计算可以吗?四、配样为什么不一次配好?还移液一次就不是很精确了,25mg到50ml不可以吗?
yankunsu
第7楼2011/01/27
进样量不对,配置标准品时,应配置不同浓度而进样量相同。如果定量环是20ul你进25ul、30、50都是20ul而且5ul、10ul、15ul、20ul在进样中并不成比例
jinp590313
第8楼2011/01/30
同意7楼观点,标准曲线是错的。应配制梯度浓度,进样量在50ul为好。
品牌合作伙伴
执行举报