气相色谱法测定水果蔬菜中17种有机氯和拟除虫菊酯类
摘要: 目的:建立毛细管柱气相色谱法测定蔬菜中17种有机氯和拟除虫菊酯类农药残留的分析方法。方法:蔬菜经均质,用乙腈提取,80℃氮吹,正己烷溶解经博纳艾杰尔的SPE小柱过滤后,再用50℃氮吹,用正己烷定容;采用VF-1MS石英毛细管色谱柱和电子捕获检测器(ECD)测定,一次进行17种有机氯和拟除虫菊酯类农药残留检测。结果:样品加标回收率在82.4%~102.5%之间,变异系数小于20%,相对标准偏差0.9%~5.2%之间,检出限在0.0001mg/kg~0.002mg/kg之间。结论:实验结果表明,该方法简便、快速、灵敏、准确,各项技术指标满足蔬菜中17种有机氯和拟除虫菊酯类农药残留检测要求。
关键词:气相色谱;蔬菜;有机氯和拟除虫菊酯类农药;残留
有机氯农药是用于防治植物病、虫害的组成成分中含有有机氯元素的有机化合物,是一种高毒高残的常用农药,最常用的为六六六和DDT等,有机氯农药中毒是指接触过量有机氯农药引起损害中枢神经系统和肝、肾为主疾病。拟除虫菊酯类农药是一类模拟天然除虫菊酯素的化学结构而合成的农药,亦称仿生合成农药。拟除虫菊酯类农药经呼吸道、皮肤和消化道侵入机体,毒性一般为中等毒性和低毒。可致神经系统兴奋性增高。近年来,由于不科学和超量使用,造成害虫对该类农药的抗药性不断增强,用药浓度在不断加大,其残留量也随之增加,从而对人类危害逐渐加重,成为农残检测中较普遍的一类农药。虽然有机氯和拟除虫菊酯类残留检测较为常规,但是由于样品基质的不同、净化的复杂等等各方面因素,在日常的检测中仍有许多的问题。
蔬菜中有机氯和拟除虫菊酯类农药残留的分析方法主要有GB/T5009.110-2003①, GB/T5009.146-2003②,NY/T761-2008③,前两种方法样品处理较为复杂,测定时间长,后一种是农业部发布的“蔬菜和水果中有机氯和拟除虫菊酯类农药多残留检测方法”本实验采用后一种方法并结合实际工作加以改动,即蔬菜经乙腈提取后少量盐析后,浓缩,80℃氮吹,经博纳艾杰尔的SPE小柱过滤后,再用50℃氮吹,用正己烷定容,采用气相色谱法VF-1MS石英毛细管色谱柱和电子捕获检测器(ECD)测定,一次进行17种有机氯和拟除虫菊酯类农药残留检测。具有快速、灵敏、准确的特点,满足蔬菜中17种有机氯和拟除虫菊酯类农药残留检测需要。
1材料与方法
1.1 仪器与试剂
瓦里安450-GC气相色谱仪,配有ECD检测器,ComPiBAL8400自动进样器,GALAXIE中文/英文工作站软件控制系统;德国博朗MR5550手提式食品调理机;FJ200高速分散均质机;弗罗里析柱④;乙腈(色谱纯)、丙酮(色谱纯)、正己烷(色谱纯)、氯化钠(140℃烘烤4h)。六六六、DDT、百菌清、三唑酮、联苯菊酯、甲氰菊酯、三氟氯氰菊酯、氟氯氰菊酯、氯氰菊酯、氰戊菊酯、溴氰菊酯等标准溶液(100ug/mL):由农业部环境监测中心提供。
1.2 样品前处理
1.2.1样品制备
取不少于1000g蔬菜样品,取可食部分,用干净纱布轻轻擦去样品表面的附着物,采用对角线分割法或四分法,取对角部分,将其切碎,充分混匀放入食品调理机,制成待测样,放入分装容器中备用。
1.2.2提取
准确称取25.0g样品放入250ml的广口试剂瓶中,加入50.0 ml乙腈,在均质机中高速匀浆2min后用滤纸过滤,滤液收集到装有5~7g氯化钠的100 ml具塞量筒中,收集滤液40~50 ml,盖上盖子,剧烈震荡1min,在室温下静止10min,使乙腈和水相分层。
经多次试验,氯化钠的使用量3~5g效果最好,如果使用量在7g以上,则在以下过滤步骤中有可能将细小氯化钠带入过滤溶液,在二次通过弗罗里析柱时需要时间更长,容易造成样品溶液的挥发,实测值偏大。室温下静止时间在30min左右所得溶液更纯净,含量更精确,若少于10min会造成所得溶液有一定的含水量,在恒温氮吹浓缩时,会有较大误差。
1.2.3净化
从100 ml具塞量筒中吸取10.00ml上层乙腈溶液,放入50ml烧杯中,将烧杯放在80±1℃水浴锅上加热,杯内缓缓通入氮气,蒸发近干,加入2.0ml正己烷,盖上铝箔待检测。
1.2.4过滤定容
弗罗里析柱的选择:选用博纳艾杰尔的SPE小柱(有机相6ml,1000mg),将弗罗里析柱依次用5.0ml丙酮+正己烷(10+90)、5.0ml正己烷预淋条件化,当溶剂液面到达柱吸附层表面时,立即倒入样品溶液,用50ml刻度离心管接受洗脱液,用5ml丙酮+正己烷(10+90)涮洗烧杯后淋洗弗罗里析柱,并重复一次。将盛有淋洗液的小烧杯置于氮吹仪上,在水浴50±1℃温度条件下,氮吹蒸发至近干,用正已烷准确定容至5.0ml,在旋涡混合器上混匀,待测。
样品一式两份做平行样。
1.3 仪器设置
1.3.1色谱条件
进样口温度,260℃;检测器温度,300℃;柱温,150℃(保持1min)→15℃/min→240℃保持1min→10℃/min→280℃保持10min,程序升温总计22 min;载气:氮气,纯度≥99.999%,色谱柱载气流速为2mL/min;进样方式为分流进样,分流比为20:1。
样品一式两份。
1.3.2色谱分析
准确吸取1.0ul标准混合溶液(或净化后的样品溶液)注入色谱仪中,以保留时间定性,以样品溶液峰面积与标准溶液面积比较定量。
1.4结果
1.4.1定性
样品中未知组分的保留时间分别与标样在的保留时间相比较,如果样品中某组分的两组保留时间与标准中某一农药的两组保留时间相差都在±0.05min内的可认定为该农药。
1.4.2计算
样品中被测农药残留量以质量分数ω计,数值以毫克每千克(mg/kg)表示,按下面公式计算。
ωmg·kg-1=
式中:
p――标准溶液中农药的含量,单位为毫克/升(mg/L);
A――样品中被测农药的峰面积;
AS――农药标准溶液中被测农药的峰面积;
V1――提取溶剂总体积;
V2――吸取出用于检测的提取溶液的体积;
V3――样品定容体积;
m――样品的质量。
计算结果保留三位有效数字。
2结果与讨论
2.1 单点校正方法
将17种有机氯和拟除虫菊酯类农药配成0.10、0.20和1.0ug/ml混合标准溶液,取样1ul在上述色谱条件下测定,做方法模板,采用单点校正的外标法,和样品组分进行比较计算。
2.2精密度与检出限
按上述色谱条件对1.0ug/ml混标平行测定5次,结果见表2。
表2 17种有机氯和拟除虫菊酯类农药
精密度试验和检出限
农药名称 相对标准偏差RSD(%) 检出限(mg/kg) |
α-六六六 2.8 0.0001
β-六六六 2.8 0.0001
γ-六六六 3.0 0.0001
δ-六六六 3.0 0.0001
p,p'-DDT 3.1 0.0002
o,p'-DDT 2.8 0.0001
p,p'-DDE 2.4 0.0004
p,p'-DDD 3.1 0.0009
百菌清 0.9 0.0003
三唑酮 0.9 0.001
联苯菊酯 1.9 0.0006
甲氰菊酯 1.8 0.002
三氟氯氰菊酯 2.8 0.0005
氟氯氰菊酯 3.8 0.002
氯氰菊酯 4.2 0.003
氰戊菊酯 4.5 0.002
溴氰菊酯 5.2 0.001 |
2.3回收率试验
在25.0g黄瓜样品中添加100ug/ml的17种菊酯农药混合标准溶液至添加浓度为0.1、0.2、1.0 ug /ml,按1.3(样品处理)的步骤处理和测定,测定结果见表3
表3 17种菊酯农药混标的回收试验(n=7)
农药名称 添加量(ug) 本底值 检测值(ug) 回收率(%) |
α-六六六 0.1 0 0.099 99
β-六六六 0.1 0 0.102 102
γ-六六六 0.1 0 0.100 100
δ-六六六 0.1 0 0.098 98
百菌清 1.0 0 0.989 98.9
三唑酮 1.0 0 0.912 91.2
p,p'-DDT 0.2 0 0.197 98.5
o,p'-DDT 0.2 0 0.188 94
p,p'-DDE 0.2 0 0.203 101.5
p,p'-DDD 0.2 0 0.205 102.5
联苯菊酯 1.0 0 0.909 90.9
甲氰菊酯 1.0 0 0.899 89.9
三氟氯氰菊酯 1.0 0 0.876 87.6
氟氯氰菊酯 1.0 0 0.874 87.4
氯氰菊酯 1.0 0 0.876 87.6
氰戊菊酯 1.0 0 0.848 84.8
溴氰菊酯 1.0 0 0.824 82.4 |
2.4色谱图
图1 17种有机氯和拟除虫菊酯类农药标样色谱图,四种666标液浓度0.1 ug/ml,四种DDT标液浓度0.2 ug/ml,其他标液浓度均为1.0ug/ml
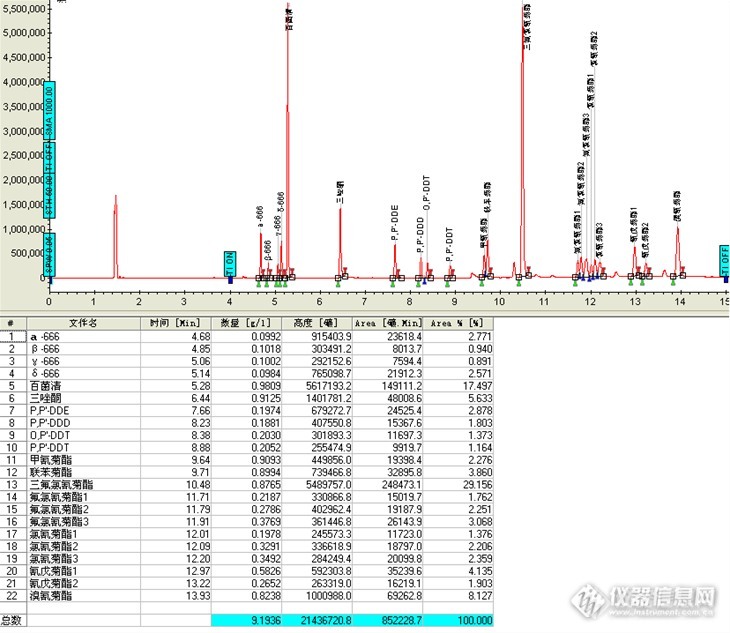
图7 黄瓜样品添加回收率色谱图 各添加浓度:四种666标液浓度0.1 ug/ml,四种DDT标液浓度0.2 ug/ml,其他标液浓度均为1.0ug/ml
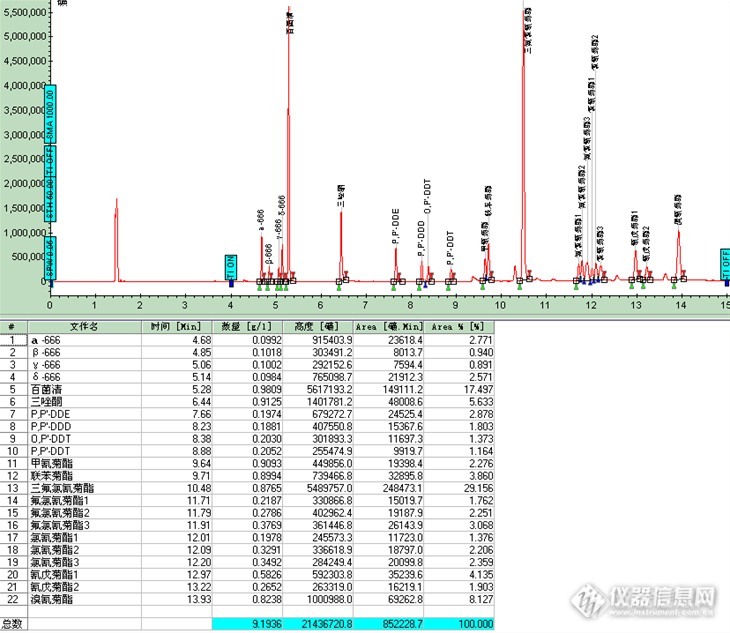
3结论
实验表明,蔬菜经乙腈提取后盐析,浓缩经弗罗里析柱后再浓缩,用正己烷定容,采用气相色谱法VF-1MS石英毛细管色谱柱和电子捕获检测器(ECD)测定,一次进行17种有机氯和拟除虫菊酯类农药残留测定。样品加标回收率在82.4%~102.5%之间,相对标准偏差在0.9%~5.2%之间,检出限在0.0001mg/kg~0.002mg/k之间。经过2008年以来我们参加河南省农业厅组织的全省蔬菜产品例行监测工作中对各种水果蔬菜检测结果表明,该方法具有简便、快速、灵敏、准确的特点,可以满足水果蔬菜中17种有机氯和拟除虫菊酯类农药残留日常检测需要。




































