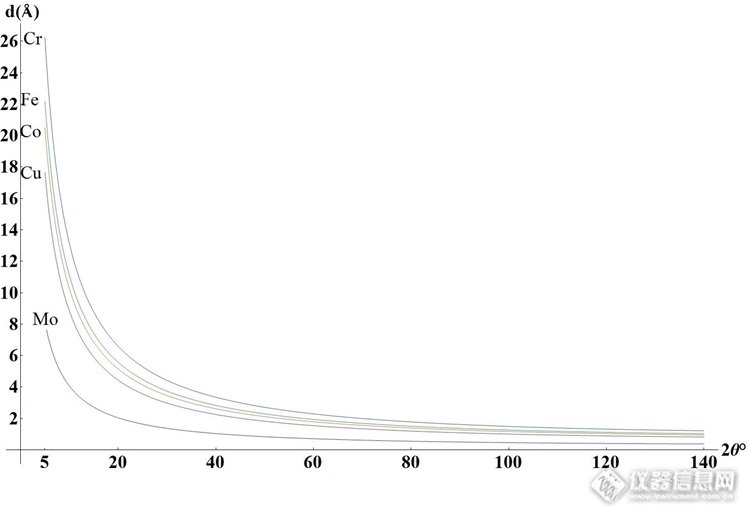
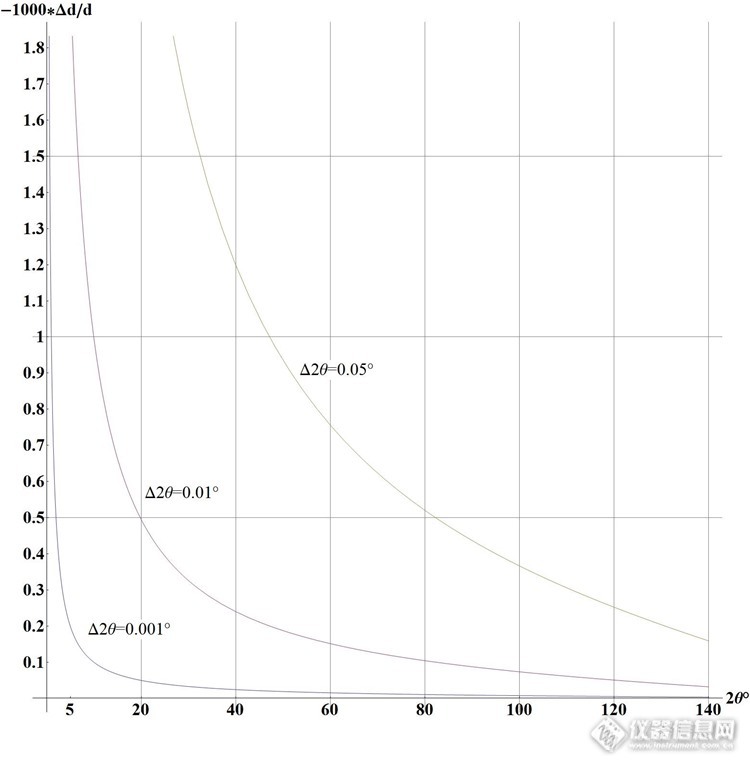
X射线衍射仪(XRD)
秋月芙蓉
第1楼2014/08/31
感谢版主的无私分享,对初学者很有帮助
iangie
第2楼2014/09/01
应该的.欢迎版友们在楼下对文章内容提问讨论.
黄石道人
第3楼2014/09/02
楼主威武,那么多标样,太牛了,有一个问题请教下,钢铁中奥氏体的标样是什么意思?用来标定奥氏体和铁素体相对含量的?
第4楼2014/09/02
都是我们老教授收集的, 485-488是老标样了, 是一块固体片片,里面包含标准含量(5% 15% 30% 2%)的奥氏体 现在在http://www.nist.gov/srm/上搜索Austenite关键字还能查到它们的certificate.
daxu
第5楼2014/09/06
的确不错,谢谢分享。事实上样品的粒度并不是越细越好,应该是5um到10um之间比较好,但很难控制。另外建议版主既然花时间写出来了,应该再加个标题序号啥的顺一顺可能读起来更有主题感
第6楼2014/09/11
谢谢意见 标题加粗了 主要是一些常识点,其实并不太有顺序.
allen_bian
第7楼2014/10/10
谢谢楼主,楼主有没有通俗易懂的书推介!
第8楼2014/10/16
没有, 中文专著我看的少。。。
wjg
第9楼2014/11/02
版主,水平高,实用
zhaosuping2011
第23楼2016/01/12
楼主你好,有个问题想问下,我的XRD衍射仪的峰偏移了,怀疑是光路有问题,想问下有解决的办法吗
品牌合作伙伴
执行举报