原子吸收光谱(AAS)
生于八零年代
第1楼2016/05/06
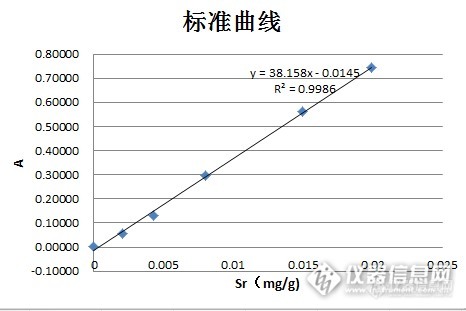
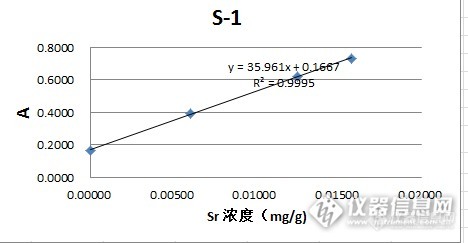
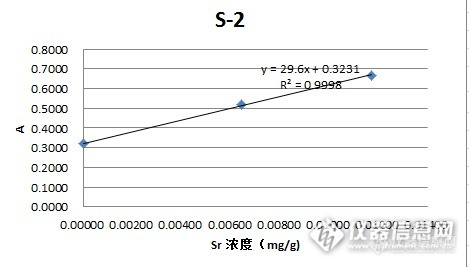
做的很不错的标准加入法,后面两张曲线图每个加标浓度能标准下最好了。楼主可否上传相关的EXCEL文件。
冰斗
第2楼2016/05/07
这个有可能是因为浓度过高造成的。如果不是什么特殊要求的话,最好把样品的吸光度控制在0.1左右,一般做加标前先估算下样品的浓度,然后加标第一点是估算出的样品浓度,后面的按倍数排列。整个分析过程的吸光度范围尽亮控制在0.04~0.4之间。
冰山
第3楼2016/05/07
样品溶液的信号值随着时间变化而产生了较大的变化一般来说是仪器的问题,但首先必须排除掉在此过程中可能发生的蒸发甚至污染等因素。发生这样的情况,就表明需要重新做标曲才能进行样品测定
yyg0506202
第4楼2016/05/09
谢谢您的提醒。
第5楼2016/05/09
估计是仪器原因多一些。AAS 测试较长时间后,吸光度发生变化是一个普遍现象。我正在考虑这样一个方法:每测10个样品,或者测试半个小时之后;重新测试标准曲线的点,如果与之前变化很大,则重新测量这个点的吸光度,根据变化系数校正曲线。希望给些建议!谢谢
第6楼2016/05/09
还有一个问题:有些文献提到使用EDTA屏蔽阳离子干扰。AAS说明书中提及的干扰主要是酸根离子的干扰。酸根离子的干扰我可以理解。我对阳离子的干扰有些疑惑,使测量结果偏大?
第7楼2016/05/09
有文献提及用EDTA是可以消除阳离子干扰,但是当达到一定浓度时,会产生络合物晶体,严重干扰分析测定,推荐使用柠檬酸铵。这个没有具体试验过,可以去试试。
品牌合作伙伴
执行举报