说明:本文最初发布于“热分析与吸附”公众号(点击打开链接),欢迎关注公众号了解更多的与热分析和吸附相关的内容。
通常通过色谱法、光谱法以及滴定法等技术来测量物质的纯度,然而在实际应用中这些传统方法具有前处理繁琐、耗时等不足。与这些传统分析技术相比,通过热分析中的差示扫描量热法(DSC)测量物质的纯度则具有样品用量少、基本不需要前处理、耗时短等优势。在本部分内容中将简要介绍通过DSC法测量物质的纯度的方法。
1. DSC法测量纯度的方法简介
根据样品和杂质性质的差异,可以采用多种方法来分析药物和小分子有机物的纯度。测定药物纯度的方法有多种,如溶解度分析法、液相色谱法、红外光谱法、紫外光谱法、滴定法等。传统分析方法比较复杂及费时,例如现有的高效液相法,在实验中需要使用大量有机试剂,易对环境造成污染。并且该方法的前处理及实验工作量大、测定时间长,对测试者的人身威胁也大。滴定法由于测试环境简单、操作带来的误差较大导致得到的数据往往不可靠。另外,由红外光谱、紫外光谱等方法只能给出峰的变化,只能得到一个相对的数据,无法得到较准确的定量数据。
与其他测定纯度的方法相比,DSC法具有以下的优点:
(1)试样用量少,一般只有1-5mg;
(2)测定时间短,通常少于1h;
(3)不需要标准品;
(4)样品制备简单,不需分离杂质;
(5)能测定物质的绝对纯度,并且在精确度和准确度上优于其它方法。
随着现代热分析技术和热分析理论的发展,差示扫描量热法用于测定物质的纯度的实例越来越多。DSC法作为一种简单、可操作性强、测试时间短的测定纯度的方法,较目前已有的测定纯度的方法优势巨大。DSC法可以在几个小时内利用很少的样品(几微克)判断出样品的绝对纯度,具有精度高、数据可靠性高等优势。
采用DSC法测量有机化合物纯度,只基于化合物的熔融过程的吸热峰,不需要任何参考标准,甚至样品的纯物质熔点也不必知道。用DSC测定物质的纯度时,试样的纯度对曲线的峰高和峰宽有明显的影响。从理论上看,高纯度且结晶完善的化合物,其DSC熔融峰非常尖锐。熔融峰变宽是估量化合物纯度的标准。纯度降低时,熔融起始温度降低,峰高度下降,熔程加宽(图1)。因此,通过简单的峰形对比也可简便地估计样品的纯度。
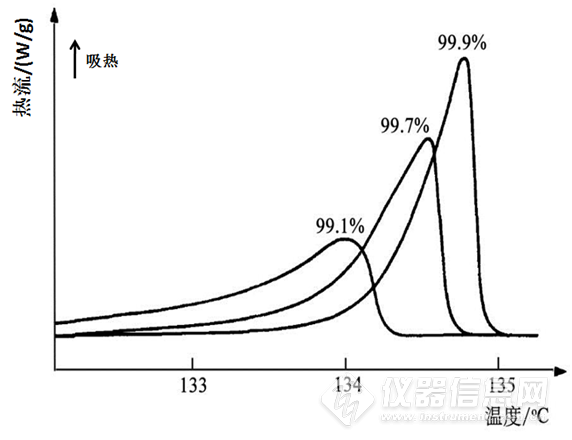
图1 不同纯度的小分子有机物的DSC曲线
当然,采用DSC测定物质纯度的过程中存在着一些假设条件和不能克服的仪器因素,有时会影响测量结果的准确性。为减少理论和实际之间的偏差,提高检测的准确性,理想的化合物试样应满足以下条件:
(1)纯度在98%以上;
(2)杂质不与主要成分起反应,不与主要成分形成共晶或固溶体;
(3)试样中的杂质与熔融的目标物质之间具有化学相似性,即在液相状态时可以互熔,并形成理想溶液;
(4)试样若存在多晶现象,必须全部转变成某一晶型;
(5)试样在熔融过程中化学性质稳定。
2 DSC法测量纯度的工作原理
DSC中常用测量纯度的方法有单峰法和多峰法。
(1)单峰法
DSC法测定物质的纯度的理论基础是共熔体系熔点降低原理。在主成分和杂质所形成共熔体系中,主成分的熔点会随杂质的摩尔百分比含量的提高而逐渐降低。从理论上看,对于纯度高并且结晶完善的化合物而言,其DSC熔融峰非常尖锐。熔融峰变宽是估量化合物纯度的标准,纯度降低,熔融起始温度降低,峰高度下降,熔程加宽。根据Van’t Hoff方程,样品熔点Tm与纯品的熔点T0差值为:
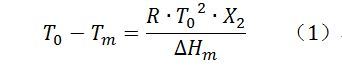
其中表示摩尔热熔热焓,R为理想气体常数,杂质的摩尔常数,为纯物质的熔点,为含杂质的材料熔点。
为得到T0与ΔH,通常引入了熔融过程中任意样品温度Ts下熔融分数F概念,即
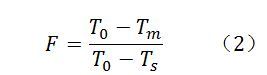
将Van’t Hoff方程代入F,得到下式:
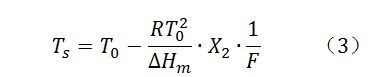
等式(3)中,以Ts对1/F图应为一直线,该直线在Ts轴上的截距即为纯样品熔点T0,从直线的斜率可求出X。实际上Ts对1/F并不呈直线关系,而是随着试样纯度的降低而更远地偏离直线。主要原因是当实验过程已经达到热平衡时,试样尚未形成理想的固体溶液,而DSC实验曲线却已开始记录,这种现象称为样品的预熔融。试样预熔融面积在试样初熔面积中占有相当比重,使DSC曲线求得的熔融部分的分数F偏低。
为了消除这种偏离信息,需进行校正处理。在数据分析时加一小块面积Sx到各部分面积Sn和总面积S中。通过线性最小二乘法拟合的方法直到Ts对1/F拟合为一条直线来确定Sx。通过选取DSC曲线中线性最好的一段进行横坐标(温度)等分,由DSC曲线得到不同的熔融温度以及所对应的热焓值,以Ts对1/F作图,求出几个Ts时的熔融分数,便可以计算出杂质含量。
图2是利用DSC对双酚A纯度进行测定得到的DSC曲线。可以由DSC曲线按照以上的方法直接计算得到杂质含量,而不需要使用高纯度的标准物质,使得实验操作误差对纯度的影响大大降低。
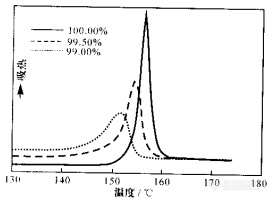
图2 双酚A纯度对DSC曲线的影响
(2)多峰法
由于单峰法测试的纯度范围比较小(>99%),而且需要校正因子来进行修正。在此基础上发展了多峰法。多峰法采用步阶式升温,在平衡条件下完成融化过程,精度比较高,而且测量范围广。常用的多峰法是双峰法,只测多峰法中的最后几个峰,因为这几个峰的面积大,面积相差也大,因此误差比较小。这种方法选用步阶升温技术,分步完成熔融过程,这样就可以把预熔过程分离开来,各个峰的总和为,这样得到的-F为一条直线,不需要修正,这种方法最大的好处是可以测定杂质量较大的物质纯度。
3 影响纯度测量结果的因素
在对DSC曲线进行纯度分析的过程中,影响纯度测量结果准确度的因素主要包括:
(1)样品量的影响
样品量大小对于测定结果的灵敏度及准确性有明显影响。较小的样品量有利于消除升温过程中样品的温度梯度,提高测定的灵敏度。但较小的样品量受外界影响而产生的误差较大,样品量存在一个最佳值。对实验结果进行二次拟合,考虑到允许的测定误差即不同样品的摩尔分子量不同,一般取2-3mg的样品进行实验。
(2)升温速率的影响
样品的升温速率是影响测定结果准确性最重要的因素。由于在升温过程中样品存在温度梯度,造成测量温度与实际温度的差别。较低的升温速率有利于提高测定的准确性,但会增加实验耗时。通过实验,在温升速率低于1℃/min对实验结果的准确性不会带来影响。
(3)坩埚的选择及预处理
为了保证测定准确性的要求,并考虑到经济条件,对一般无特殊腐蚀性样品的测定,一般铝质坩埚能满足要求;由于纯度测定温升速率较慢,使用常用的半密封坩埚会造成熔融或半熔融状态下样品的挥发,因此必须使用密封坩埚,这对测定易挥发性样品尤为重要。对高精度测定实验,坩埚在使用之前必须进行加热预处理。
(4)保护气流速影响
在实验过程中通入N2、He气的主要作用是消除样品热裂解而产生的有害气体,保护气流速对测定准确度的影响不是关键因素,一般选择20-40ml/min。
(5)样品粒度大小的影响
当样品的粒度过大时,由于热传递变慢而产生热梯度,导致峰形变宽。如对大颗粒进行研磨,容易因静电作用而发生团聚,因此需要较多的能量才能将其熔化。一旦熔融则速度加快,其熔融峰比原来团聚小晶体的熔融峰更尖锐。颗粒较小则有较多的晶体缺陷,试样处于较高的能量状态,导致峰高降低,峰形变宽,测得的纯度偏低。
另外,为了获得具有可比性且重现性好的实验数据,对于在不稳定环境下的易吸潮样品,通常在温度控制程序的起始平衡温度前加入一段样品等温过程,以消除样品的自身影响因素。
由于以上影响因素的存在,可以通过不确定度分析来评价测量结果的可靠性。
综合以上分析,用DSC法测定物质的纯度十分简单,具有准确度、精密度高,重现性好,操作简单,不需分离杂质等优点,完全可以作为测定有机化工品、药品纯度的常规检验方法。据估计,有75%以上的结晶有机化合物可用差示扫描量热法进行纯度测定。但由于影响热分析方法的精度的因素较多,如仪器因素、样品在纯度、稳定性及晶型等方面的因素等,因而热分析法在实际使用时有一定局限性,不能作为药物纯度判断的唯一标准,但可用于确证和支持其它分析方法纯度测定的结果。



































