


2.1 材料与试剂
安赛蜜、糖精钠、苯甲酸、山梨酸、脱氢乙酸、咖啡因、阿斯巴甜标准品纯度均不低于99.5% 上海阿拉丁公司;甲醇(色谱纯)、磷酸二氢铵(分析纯)、氨水(分析纯) 上海国药试剂公司;实验用水为二次蒸馏水。2.2 仪器与设备
iChrom 5100色谱系统,配D5115二极管阵列检测器(DAD) 大连依利特公司;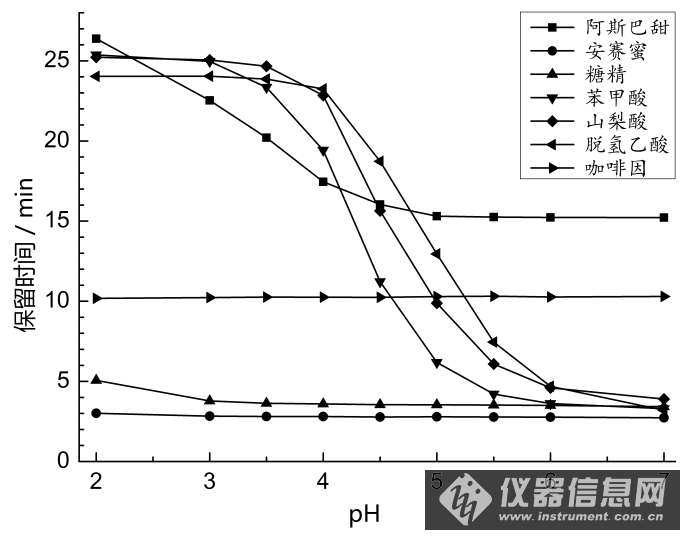
流动相pH对7种目标物保留时间的影响(流动相为甲醇/缓冲液=20/80)
.
糖精钠和安赛蜜都是强酸盐,相当于非常弱的碱,咖啡因本身就是极弱的碱,因此他们在弱酸性范围内存在形式不会有显著变化。只有在pH小于2之后,其保留时间才会水pH显著变化,但是通常色谱柱不能承受强酸,因此对pH小于2的情况不予考虑。阿斯巴甜是两性物质,在等电点附近呈分子内电离的形态,是保留最弱的。当pH增大或者减小时,其电离程度都是减小,保留时间都会表现为增大,但pH大于8是通常色谱柱不能承受的,因此也不予考虑。苯甲酸、山梨酸、脱氢乙酸都是羧酸,pKa均在5左右,因此其存在形式在pH从4到6变化时会显著不同,其保留时间在这个pH范围内也是变化最为显著的。pH减小时,羧酸形式存在占主导,因此其保留时间变大;pH增大时,盐形式存在的比例变多,因此保留时间减小;并且对于酸性强度不同的物质,其变化幅度和变化发生的pH范围也是显著不同的。相关理论在众多色谱专著中均有讨论(参见:Lloyd R.Snyder,Joseph J.Kirkland ,John W.Dolan,(译者:陈小明 唐雅妍),现代液相色谱技术导论,人民卫生出版社,2012.)。
上述讨论也说明,对于酸类物质的色谱分析,一定要精确控制流动相的pH。有些标准中对流动相pH未作明确的说明,这是极为不妥的,试剂的微小差异就会导致分析结果显著不同。例如GB 5009.28-2016中只规定了流动相加入乙酸铵,但实际购买的乙酸铵试剂pH可在6.5~7.5范围内变化,有时候就会导致苯甲酸与糖精钠的峰位置前后互换。
在明确了上述变化规律之后,流动相pH选择的问题也迎刃而解:显然在pH=5.5附近对于分离是最为有利的,不仅各目标物分离度较高,而且总的分析时间不太长,只需约15分钟。需要指出的是,由于pH计往往存在误差,实际配置缓冲液时并不一定按图中数值,而需要根据实际情况进行微调。若实测中发现脱氢乙酸与咖啡因分离度较低,说明配置的缓冲液pH略有偏低,应适当调高pH(通常调高0.1单位即可改善);若实测中发现脱氢乙酸与山梨酸分离度较低,说明配置的缓冲液pH略有偏高,应适当调低pH(通常调低0.1单位即可改善)。
在上述讨论的最佳流动相条件下,标样和实际样品的分离效果都很好,见下图:
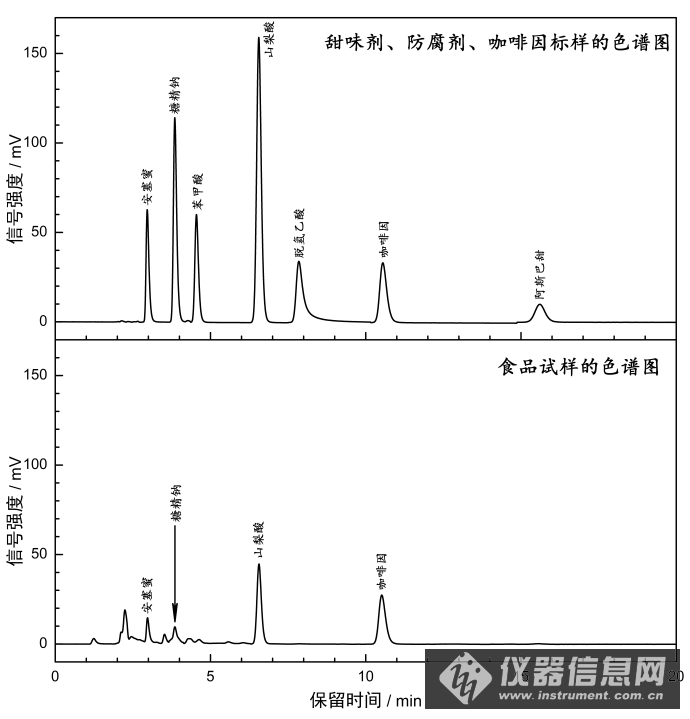
标样和实际样品的色谱图(流动相为甲醇/缓冲液=20/80,缓冲液为0.1M磷酸二氢铵溶液用氨水调节至pH5.5)
.
另外要补充说明的是,虽然标准方法中使用的缓存盐常常是乙酸或者甲酸体系,但实际上并不是最佳选择。因为有机酸本身会产生一定的背景吸收,在低波长下尤其明显。而磷酸盐体系的背景吸收要小得多,这对减小背景干扰和基线漂移是有利的。
3.2 检测波长选择
7种目标物的的吸收波长各不相同,用DAD检测器可较好解决问题,并且有助于通过吸收光谱图定性。若没有配备DAD检测器,可用一般可变波长紫外检测器的“波长时间程序”功能。
通过较高浓度标样测得7种目标物的色谱-光谱图如下,其紫外吸收谱图和最大吸收波长可供参考。

需要说明的是,糖精钠和阿斯巴甜的最大吸收波长都在较低波段,易受到背景吸收和样品基体的干扰,因此实际选用的是210nm进行检测,灵敏度虽然有所降低,但干扰也明显减轻。实际应用中发现,少数情况下,糖精钠仍然会受到基质干扰,此时可进一步将其检测波长降低到215~220nm,基质干扰基本上就减小到可忽略的程度了。
另外,不同文献中报道的各种目标物的最大吸收波长有一些出入,这一方面与不同仪器的波长误差有关,更重要的是因为目标物的离解程度随pH变化,不同pH下测得的最大吸收波长会有明显差异。
3.3 样品前处理
本方法参考现有方法,采用了锌盐沉淀的吸附和絮凝作用来净化除杂,但是未使用亚铁氰化钾和乙酸锌,也未采用三氯乙酸沉淀蛋白。因为这几种物质都有末端吸收,在接近210nm处产生的紫外吸收较为明显,而其色谱流出的时间又都与安赛蜜、糖精钠等物质接近(例如,三氯乙酸的流出时间与糖精钠几乎相同)。本方法采用氨水和硫酸锌生成氢氧化锌絮状沉淀,其吸附与絮凝效果与亚铁氰化钾-乙酸锌体系较为接近,同时不引入干扰离子。
对于含油脂较多的试样,本方法仍采用正己烷萃取除脂,与常规方法基本一致。
3.4 方法验证
对方法的重现性、线性范围、加标回收率进行了验证,详细数据较多,这里从略,主要结果如下:
(1)标样7次重复测定的重现性很好,RSD均小于1%。但样品处理的重现性略差,同一样品匀浆后分别进行7次净化和测定,RSD在1%~3%。
(2)7种目标物在1~50mg/L范围内线性相关系数均大于0.999,以取样1.0g计,测定范围相当于0.025~1.25g/Kg。考虑到添加剂的实际使用情况,未对更高含量水平进行验证。
(3)按三倍信噪比估算,检出限均低于0.1mg/L,远低于常规检测范围,故未进一步对验证检出限。
(4)进行了1g/Kg和0.1g/Kg两个水平的加标实验,阿斯巴甜的回收率略低,分别为94.1%和95.7%,其余目标物的回收率均在98%~101%之间。推测可能是阿斯巴甜容易被氢氧化锌沉淀吸附,导致回收率略低。
.
4、结论
(1)使用C-18色谱柱,使用甲醇与pH=5.5的磷酸盐缓冲液按20/80比例作为流动相,可以在等度洗脱条件下实现 安赛蜜、糖精钠、苯甲酸、山梨酸、脱氢乙酸、咖啡因、阿斯巴甜等7种物质的完全分离,分析时间约15分钟。
(2)基于上述色谱分离方法,可以实现7种常见食品添加剂的同时测定,结果较为准确,可以用于生产实践。

































