XRD
第一章XRD概述
XRD分析是一种利用X射线在晶体中发生的衍射现象进行物相鉴定的现代晶体学分析方法。通过X射线在晶体中发生特定的衍射进而形成XRD谱图。对比数据库的谱图信息我们可以对样品定性及半定量分析。[]
其实晶体具有周期性的点阵结构,又因为晶体原子距离和X射线波长在是同一数量级所以X射线会与原子产生作用而改变方向形成X射线散射,原子形成散射波,这些散射波互相干涉,导致某些方向互相加强而某些则相互减弱。所以只有在特定方向上互相加强才会存在衍射斑点。衍射线的强度则是取决于晶胞原子的种类数目和分布。通过布拉格方程的建立,我们也可以明白X射线衍射主要功能是确定晶体的物相结构和元素组成及含量,称之为X射线晶体结构分析和X射线波谱分析。所以X射线衍射既可以定性分析又可以定量分析。
第二章定性分析
由于每种晶体都有特定的结构和点阵参数,所以不同的晶体物质和X射线衍射图谱之间有特殊对应关系,所以当波长合适时满足布拉格方程(式1),就会产生相应谱图信息。
2dsinθ= nλ (1)
式中,d值由晶胞参数所决定, θ是衍射角,λ为入射波波长,n为自然数1、2、3……
所以晶体的每一衍射方向都必然和一组间距为 d 的晶面组相联系。
定性分析主要目的是判定物质中所包含的晶体组分并且可以判断出晶体物质的结晶状态。定性分析的最重要的步骤就是从d、θ等数据查的PDF卡片(也称为JCPDS卡片)信息,以确定其物相。
康乐等人[]制备了不同的催化剂材料并对其进行XRD测试如图1所示。
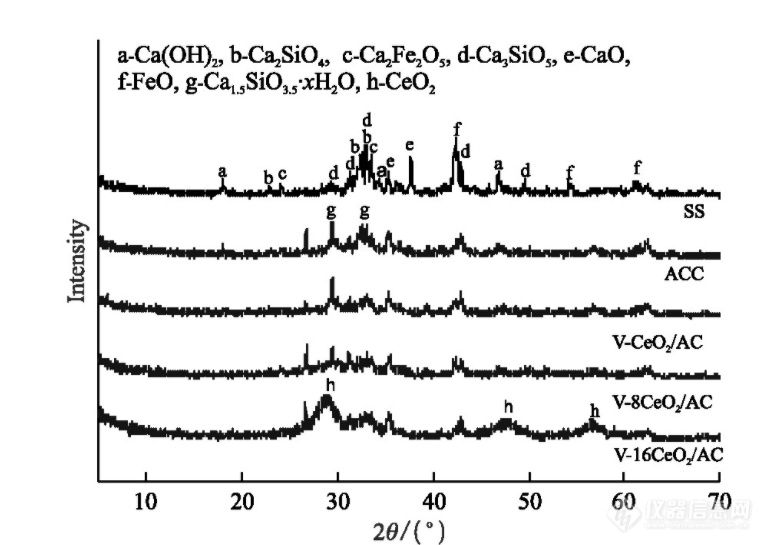
图1 合成样品的XRD图[2]
晶体主要是矿物相,查找PDF卡片可以获得以下信息,Ca(OH)2 (2θ = 18.11°, 34.35°, 46.88°, JCPDS No. 44-1481), Ca2SiO4 (2θ = 22.89°, 32.57°, 33.04°, JCPDS No. 86-0399), Ca2Fe2O5(2θ = 24.21°, 33.52°, JCPDS No. 19-0222), Ca3SiO5 (2θ = 29.46°, 31.26°, 33.02°, 33.60°, 42.95°, 49.63°, JCPDS No.84-0594), CaO (2θ = 35.42°, 37.81°, JCPDS No. 74-1226), FeO(2θ=42.47°, 54.28°, 60.98°, JCPDS No. 77-2367). 水合硅酸钙(Ca 1.5 SiO 3.5 · x H2 O,JCPDS No.33-0306),其峰值位置为2 θ = 29.46°,32.56°。
由此也可以得出XRD不仅可以确定单一物相,在多相化合物中通过对比卡片三强线d值也可以推断其他存在的物相,但是定性分析不能确定所有存在的相所以也不能判断出某个相是否存在。
第三章定量分析
定量分析目的则是确认某物相在物质中的含量,物相衍射线的强度(式2)或相对强度与物相在样品中的含量有关。常见的定量分析方法有:外标法、内标法、基体冲洗法(K值法)。前提都在假设了被测物相中的晶粒尺寸非常小且各物相都混合均匀且晶粒无择优取向。所以和实际情况有所偏差,应考虑以上误差。
I=G·C·A(θ)·V (2)
其中 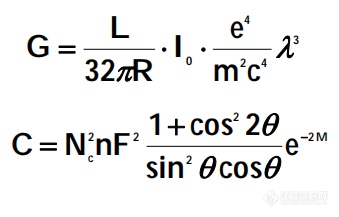
菅豫梅等人[]利用K值法快速计算硬质合金中不同钴相结构的质量分数。
Kαβ=Kα/Kβ=7.29/6.47=1.127 (3)
Wα=Aα/(Aα+KαβAβ)×100%=Aα/(Aα+1.127Aβ)×100% (4)
Wβ=100%-Wα (5)
式中,Kα代表PDF卡片中2θ角度为44.227°的α-Co(111)衍射峰所对应的RIR值;Kβ代表PDF卡片中2θ角度为47.393°的β-Co(101)衍射峰对应的RIR值;Aα代表2θ角度为44.227°的α-Co(111)衍射峰面积;Aβ代表2θ角度为47.393°的β-Co(101)衍射峰面积;Wα代表α-Co质量分数;Wβ代表β-Co质量分数。
最终计算不同牌号合金钴相结构质量分数的测试结果如下表2所示。
表2 牌号合金钴相结构质量分数结果表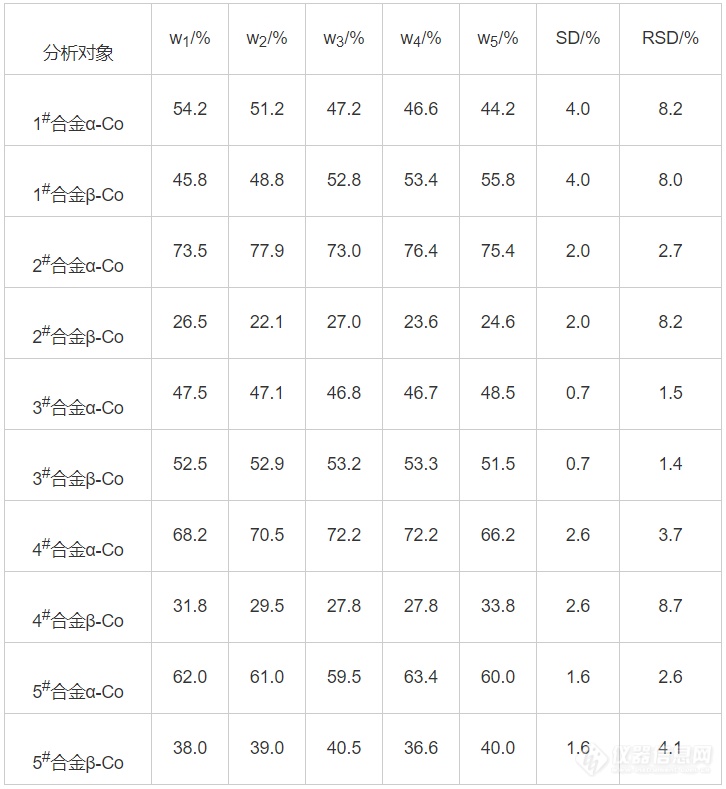
综上可以得出,相对标准偏差均小于10%,可以应用于生产分析。
由此我们可以得出,晶体物质XRD定量分析计算方法有很多种,但每一种都需要在多次重复测试结果下计算相对偏差以得到准确的结果。其定量分析与峰面积有关。
第二章物相晶体结构分析
晶体结构分析也要用到布拉格方程(式1),其次面间距和点阵参数存在以下关系(式2)d为(hkl)晶面面间距,而d满足式1,由此可以推断出晶体结构。如图1所示。
 (6)
(6)
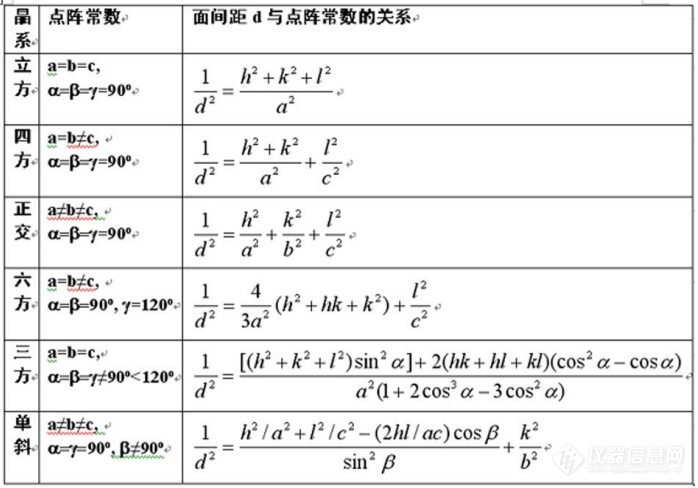
图1 面间距和点阵参数、面指数对应关系
时米东等人[]为探究催化剂ZrO2-SnO2的制备参数对其晶体结构的影响,采用不同锆锡原子比、焙烧时间等变量基于XRD探究其晶体结构变化。如图2、3所示。其中图3为Zr:Sn=3:1的500℃焙烧2~10 h的氧化物。
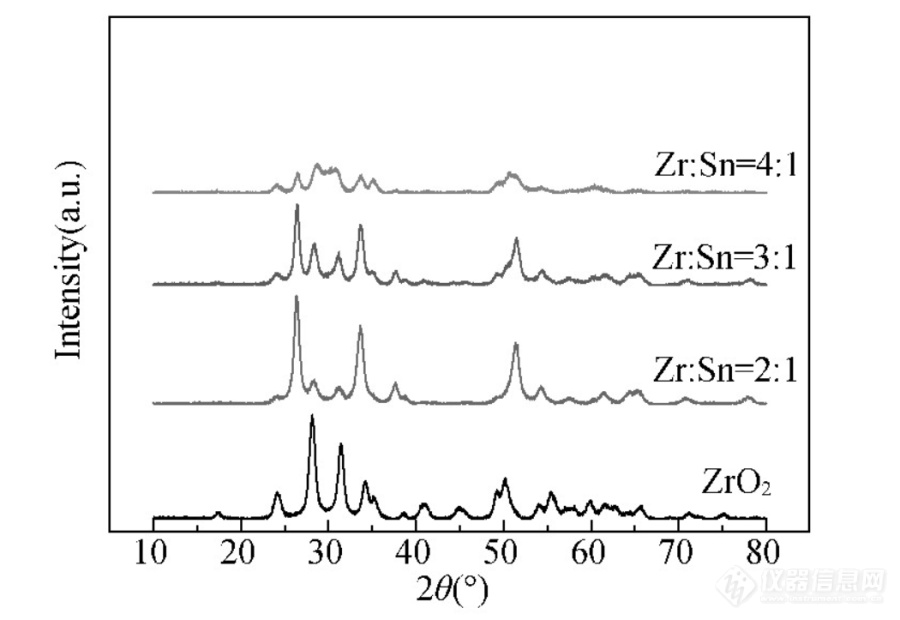
图2 不同锆锡比下氧化物的XRD图[4]
在2θ=10~80°范围,纯ZrO2为典型的单斜晶系;加入锡之后,ZrO2仍表现为单斜晶系,而SnO2则是四方晶相并不是单斜晶系。所以Sn加入并没有改变ZrO2的晶系。随着锆含量的增加,ZrO2(-111)和(111)晶面的特征衍射峰间距缩小,峰型由尖锐变得宽泛,说明SnO2对ZrO2晶体产生了影响,晶粒尺寸逐渐减小。另外,ZrO2(-111)和(111)晶面的衍射角分别右移和左移,说明锡插入到了Zr O2晶格。
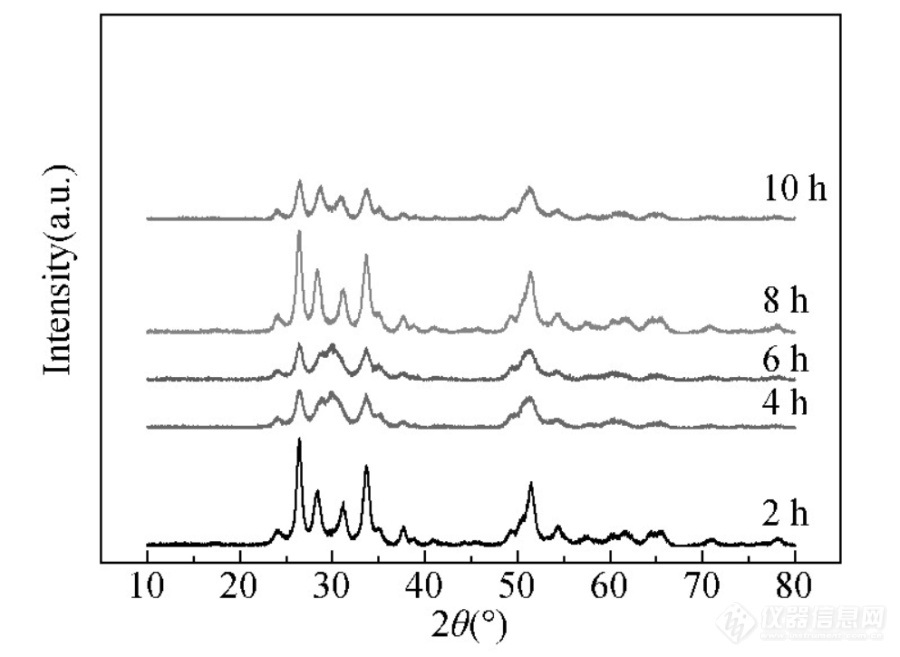
图3 不同焙烧时间下氧化物的XRD图[4]
从图3中可以看出焙烧2 h时,Zr O2和Sn O2分别表现为单斜晶系和正方晶系。焙烧4 h时,氧化物中锆元素有单斜晶系、斜方晶系Zr O2和立方晶系Zr2O;锡元素有正方晶系、斜方晶系的Sn O2和Sn O。焙烧6 h时,Zr O2表现为单斜晶系、斜方晶系和正方晶系,Zr2O表现为立方晶系;SnO2表现为正方晶系和斜方晶系,SnO为正方晶系。焙烧8h时,Zr O2为单斜晶系,SnO2为正方晶系和斜方晶系,Sn O为斜方晶系。焙烧10 h时,Zr O2为单斜晶系,Zr2O为立方晶系,Sn O2为正方晶系,Sn O为斜方晶系。所以随反应时间的延长,晶体结构逐渐单一化。
由此可以得出,当判断晶体结构是否改变时可以使用XRD测试原样和现样以及插入样。得到三种晶相进而判断,并且根据式(2)和图1以及计算软件的分析可以由θ推导出晶体面间距从而推导出晶体的结构。并且由图3结论可以衍生出若反应的时间越久那么晶体逐渐单一化。从中也可以得出XRD主要影响因素是测定的材料本身。制样时要磨细样品并用适宜的压样。
总结:XRD可以定性分析也可以定量分析,定性分析只需要通过θ利用布拉格方程从而计算出d,跟PDF卡片比对从而可以判断出含有哪些物相和物相的晶相,但是定性分析不能确定某物相不存在,所以还要进一步结合其他分析方法。而定量分析中有许多计算方法,不同方法对应不同情况,如当物相数大于2,并且各物相吸收系数不同时可以用内标法。最后XRD晶体结构分析,也可以用于定性或定量分析,并且根据θ推出d结合软件推导出hkl进一步判断晶系。还可以得出当反应时间达到一定程度时晶体逐渐单一化,所以影响XRD结果的主要因素是测定的材料本身。



































