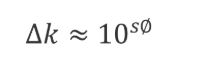仪器信息网APP
选仪器、听讲座、看资讯
立即体验
APP内打开
回版面
1
12
1
拍砖
举报
取消
发布
当前位置:
仪器社区
>
基础知识专区
>
分析化学
>
帖子详情
反相色谱中的“溶剂效应”
禹飞乐
2022/11/30
私聊
分析化学
峰形问题是反相色谱分析中最常遇到的问题之一。造成峰形问题的原因有很多,
本次我们重点讨论在反相色谱中由于稀释剂与初始流动相的差异造成的峰形问题,也就是常说的“溶剂效应”及其内在原因和应对方法。
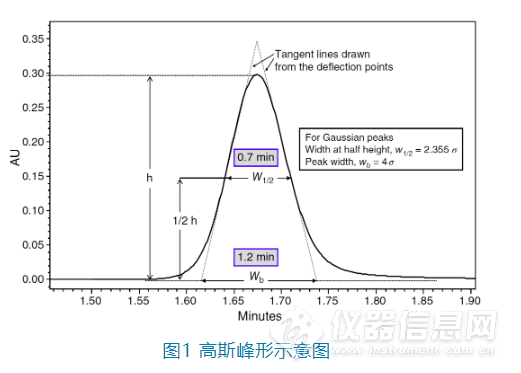
1. 为什么我们需要好的色谱峰形?
色谱分析是一种非常追求准确度的分析手段,理想的峰形是高斯(Gaussian)峰形,也就是呈正态分布,左右对称。但是在实际情况下,色谱峰或多或少都会有点前延(fronting)或者拖尾(tailing),甚至完全不成单峰等,这样
不仅影响分析人员看到图谱时的心情,也会降低色谱峰的灵敏度、峰面积的积分准确度以及色谱峰之间的分离度
。
2. 什么是理想的样品稀释剂?
在分析HPLC中,往往建议稀释剂为起始流动相(但在制备HPLC中,使用比流动相更弱的稀释剂更利于更大的进样体积),以避免对分离分析产生不利影响。但是在实际的工作中,样品的预处理往往使得其稀释剂和起始流动相有了很大的差异,也有一些分析方案中会建议通过蒸发除去样品预处理中的溶剂后再用起始流动相溶解样品。但是这种额外的步骤很费时间,甚至超过了HPLC分析的时间,只有在万不得已的时候才选择。也有基于其他考虑(如溶解性、样品稳定性等)而选择和流动相不一致的溶剂作为稀释剂。但是
不管是何种稀释剂,只要能稳定溶解足够的样品,且对峰形等不会产生不利影响,就是理想的稀释剂
。
3. 溶剂效应的原因有哪些?如何应对?
3.1 稀释剂与流动相的洗脱强度不匹配
当稀释剂的洗脱强度强于起始流动相时,也就是有机相(%B)的比例高于流动相时,我们可以估算出保留的改变(Δ
k
):
其中
S
和每种溶质的特性有关,
S
≈ 0.25 MW
0.5
,MW为分子量。如果我们选择了一种典型的400 Da的小分子,当B%变化10%(
即
Φ
=0.1
)时
,S
≈ 0.25×400
0.5
=5,Δ
k
≈ 10
5×0.1
≈ 3.2。这意味着B相的10%的增加或者降低会导致三倍的的保留因子K的降低或者增加,也就是常说的“三倍规则”。如果一个样品的稀释剂比流动相强,进样的溶液在跑过柱子的过程中,它会和流动相以相同的流速移动,但是其中的样品分子的移动速度会比溶解在流动相时更快,进样成分会被流动相逐渐稀释直到和流动相完全一致。我们把进样的液体形象地称为“样品塞”,紧跟“样品塞”的流动相会稀释它,因此尾部的溶质分子的前进速度会慢下来,以正常的洗脱速度前进。
虽然这些过程发生的速度很快,但是如果稀释剂足够强、进样体积足够大,还是会有明显的峰展宽
。
Dolan作了图来更加直观地说明这种现象,在图2中每个水平放置的长条代表充满流动相(用小圆黑点表示)的色谱柱。在上面三个例子中,进到流动相的样品用棋盘格表示,随着时间的流逝从状态A逐步变到状态B和C,样品流经色谱柱,发生了稍微的峰展宽。在下面三个例子中,样品溶解在更强的溶剂中(斜线表示),A'为刚进样后的状态,“样品塞”占据的体积和A一样,因为进样量相同;在B'中,由于“样品塞”里的溶剂比流动相更强,溶解其中的样品分子会以比正常情况更快的速度通过色谱柱,与此同时尾部被流动相稀释的部分溶质分子的通过速度降低至正常速度(棋盘格部分),这样即使“样品塞”更窄(也就是进样量更少),由于前后端的稀释,样品扩散的空间也会更大;在C'中,“样品塞”基本都被稀释了,但是峰展宽一直在发生,直到所有样品都溶解在流动相中。
在某些更极端的条件下,稀释剂的强度太大还可能导致色谱峰出现裂分甚至完全不成峰形。
当由于稀释剂由于和流动相的溶剂强度不匹配而造成峰形异常时,该如何应对?
1. 降低稀释剂的洗脱强度;
2. 降低样品的进样体积。进样体积越小,样品就越容易被流动相稀释掉。Dolan实验室尝试过在反相流动相中进样1 μL以甲苯为溶剂的样品时,可以得到满意的结果。
但是稀释剂与流动相的洗脱强度不匹配并不能解释所有情况
。比如Tseing和Roger发现分析二羟基苯的异构体时,不管是将甲醇作为稀释剂,水作为流动相还是水作为稀释剂,甲醇作为流动相,最终都得到双峰,但是当稀释剂和流动相一致时,就可以得到单峰;Zapata和Garrido也发现在分析叶绿素时,当使用丙酮-水(69:31)为稀释剂,甲醇-水(95:5)为流动相时,虽然两者洗脱强度一致,但是叶素绿的峰为多峰。经过研究后发现这和稀释剂与流动相之间的
黏度不匹配
有关。
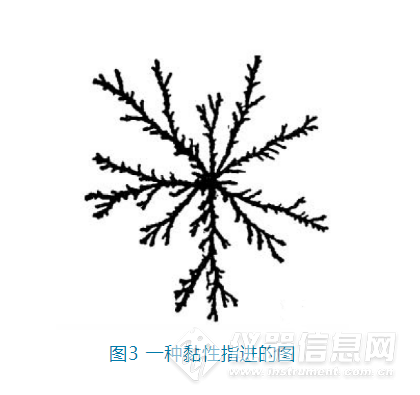
3.2 稀释剂与流动相的黏度不匹配
当稀释剂与流动相之间的黏度不一致时,会发生黏性指进(viscous fingering)这种流体力学不稳定现象。也就是低黏度的溶剂会如伸出手指一般向高粘度渗透,
同时这种渗透是随机的,不可预测
。如图3,黑色部分就是低黏度的溶剂,在更高粘度的背景溶剂中如树枝般随机“生长”。
在色谱分析中,稀释剂与流动相的黏度不匹配对谱图的影响不仅与稀释剂与流动相黏度的不匹配程度有关,也与样品溶质及样品溶剂的迁移速度有关。当黏度差异相同时,样品的溶质与溶剂在越短的时间内洗脱,且洗脱时间越接近时,对峰形的不利影响就越明显,这种现象在体积排阻色谱(SEC)中比较常见。
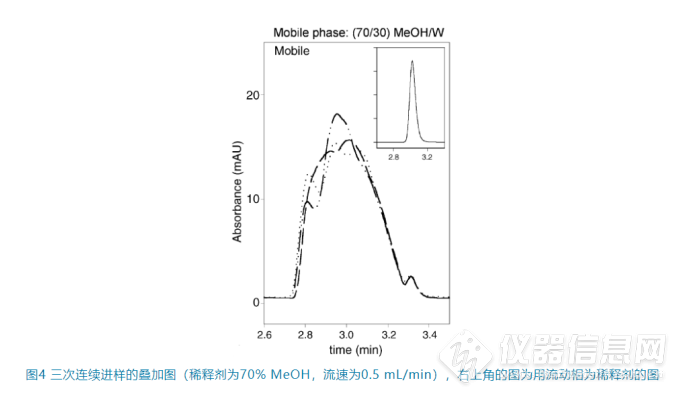
正如上面提到的这种“黏性指进”是随机的,因此
往往连续进样时峰形不能重现(如图4),这也是判断到底是黏性不匹配还是上面所说的洗脱强度不匹配的重要依据
。
当由于稀释剂由于和流动相的黏度不匹配而造成峰形异常时,该如何应对?
1. 尝试用初始流动相稀释样品,但这往往是正确的废话,因为很多情况是不能或不便用初始流动相稀释样品;
2. 降低进样量;
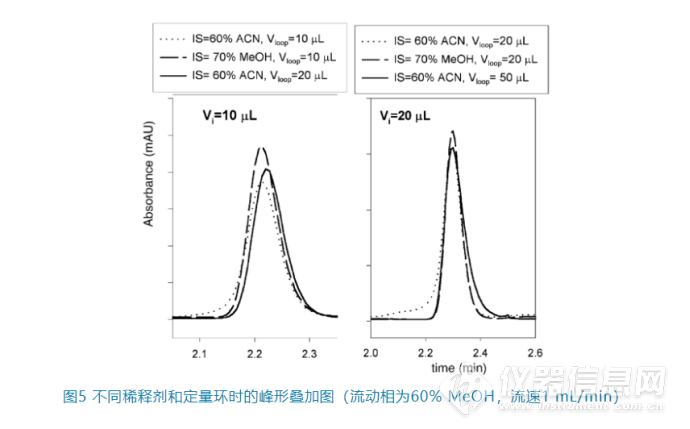
3. 选择更大的定量环,也就是增强在定量环中的预混,如图5。同样进样10 μL或者20 μL,当增加定量环的大小(从10 μL到20 μL或者从20 μL到50 μL)时,峰形可以明显改善。
4. 升高温度,可以在某些区间内拉平稀释剂和流动相之间的黏度差异,从而减少其不良影响,这在体积排阻色谱(SEC)中很常见;
5. 在半制备或者制备色谱中,常常增大进样量以提高效率,有时候由于溶解性问题也让使用起始流动相作为稀释剂不太现实,为了降低稀释剂和流动相由于黏度不匹配而造成分离的不良影响,强烈建议调节两者的黏度,一种可能的解决方案是在低黏度的一方中加入极少量的高黏度溶剂,比如DMSO或者环己醇。
3.3 稀释剂与流动相的pH不匹配
我们在分析可离子化的化合物时,常常会基于分析物的
p
K
a
选择不同pH的流动相,以实现最佳的分离效果。在很多情况下,样品稀释剂和流动相的pH并不一致,甚至相差好几个单位,比如样品在起始流动相条件下不稳定时,首先就应该考虑样品的稳定性。
当稀释剂与流动相的pH不一致时,稀释剂在流经色谱柱时,会改变周围环境的pH,从而可能改变分析物的电离状态进而影响其保留行为
。
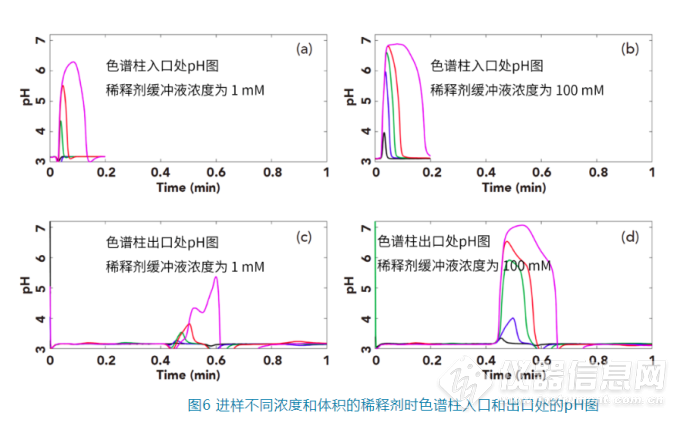
为了更加直观地了解当样品稀释剂对流动相pH的影响,Stoll在HPLC系统中设计了一个pH的在线监测系统,可以实时测定色谱柱入口和色谱柱出口的pH值。实验中选择的苯甲酸为分析物,样品稀释剂为乙腈:1 mM或100 mM磷酸盐(pH 7)=13:87,流动相为乙腈:100 mM磷酸盐(pH 3.2)=23:77,色谱柱为50×4.6 mm的C18柱,流速为2 mL/min(其他详细色谱条件见参考文献[6])。图(a)为稀释剂中缓冲剂为1 mM pH 7的缓冲盐时,不同进样量时色谱柱入口处的pH随时间的变化图;图(b)为稀释剂中缓冲剂为100 mM pH 7的缓冲盐时,不同进样量时色谱柱入口处的pH随时间的变化图;图(c)为稀释剂中缓冲剂为1 mM pH 7的缓冲盐时,不同进样量时色谱柱出口处的pH随时间的变化图;图(d)为稀释剂中缓冲剂为100 mM pH 7的缓冲盐时,不同进样量时色谱柱入口处的pH随时间的变化图。其中蓝、紫、绿、红、粉曲线分别是进样量为1 μL、5 μL、15 μL、30 μL、100 μL的pH曲线图。
在色谱柱入口端:
从图(a)可以看出,即使稀释剂中的缓冲剂为1 mM的磷酸盐,当进样量为30或100 μL时也会在流动相中产生pH>5的区域,而
当稀释剂中的缓冲剂为100 mM的磷酸盐时,见图(b),即使只进样5 μL,也会让流动相中产生pH值到6的区域
。
在色谱柱出口端:
我们可以看到其最大的pH值比色谱柱入口端监测的数据要低,这应该主要和样品的稀释剂的扩散和流动相对其的中和有关。当稀释剂中的缓冲剂浓度为1 mM时,如图C,样品稀释剂在色谱柱中充分扩散和被中和,监测到色谱柱后的pH基本都低于4,除非进样量达到100 μL时,才有少量的区域pH高于4;而
当样品稀释剂浓度为100 mM时,如图6D,当进样量为15 μL,30 μL和100 μL,会让色谱柱中很大的区域的pH都大于5
。
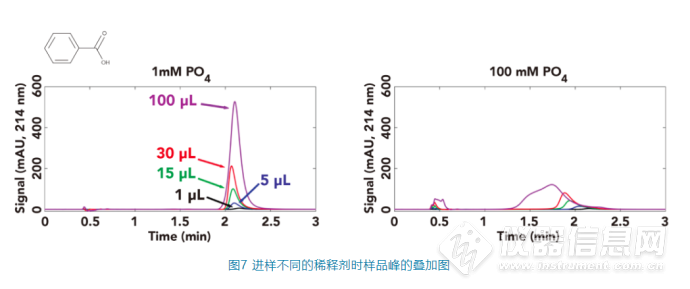
而苯甲酸的p
K
a为4.2,当色谱柱中的pH值由流动相的3.2升高时,让苯甲酸的电离程度也提高,从而让其保留时间降低,当pH值的变化比较剧烈时(如稀释剂的磷酸盐浓度为100 mM,进样量为15 μL或30 μL),色谱峰的保留时间显著减少,同时峰的拖尾变得更加严重,而当进样量为100 μL时,色谱峰会出现严重的变形和裂分现象,如图7。
综合图6和图7可以看出,分析物的出峰行为和稀释剂对流动相pH的影响情况出现了非常强的相关性。我们可以看到当稀释剂与流动相的pH不匹配时,特别在稀释剂中缓冲液的缓冲能力比较强、进样量比较大的时,对于p
K
a在特定范围内的分析物的出峰行为产生不可忽视的影响。相对应的,
降低稀释剂的缓冲能力、减少进样体积以及让稀释剂对流动相pH的影响范围尽量避开所关注的分析物的p
K
a都是有效的方向
。
全文总结
在液相色谱分析中,“溶剂效应”指由于样品的溶剂(稀释剂)与初始流动相的差异而对分析物的色谱行为造成的影响,但是其具体原因却不能一概而论。本文综合了现有的一些研究,总结了三种不同原因造成的溶剂效应,包括
洗脱强度不匹配
、
黏度不匹配
和
pH不匹配
(当然,还有可能有其他的不匹配因素导致的溶剂效应,本文仅作抛砖引玉,如有错误或者遗漏,请在评论区指出)。当遇到具体的问题时,需要具体分析找出原因,并针对性地进行改善。
参考文献
[1] Michael W. Dong HPLC and UHPLC for Practicing Scientists, 2nd Ed.
2019
, Wiley;
[2] J.W. Dolan,
LC-GC North Am.
22 (2004) 26;
[3] S. Keunchkarian et al.
J. Chromatogr. A
1119 (
2006
) 20;
[4] C.B. Castells et al.
J. Chromatogr. A
805 (
1998
) 55;
[5] M. Czok et al.
J. Chromatogr.
550 (
1991
) 705;
[6] D.R. Stoll et al.
LCGC Europe
33 (
2002
) 74.
相关话题
1
分析仪器为什么要接地?1
2
测生物碱含量
3
想寻找无紫外吸收化合物衍生案例
4
氧化锆氧分析仪
5
关于弱阴离子SPE小柱的原理问题。
+关注
私聊
zyl3367898
第1楼
2022/12/03
应助达人
没想到溶剂对峰型有这么大影响。
0
发表回复
近期热榜
仪器信息网“仪友会”招募令
科学仪器品牌联合“仪器心得”征文活动
【生活中的仪器检测】有奖征文
LC-MS实验瓶颈的突破与优化
热门活动
宝藏核磁实验室的得力助手征文活动
【售后专场--招聘会】
猜你喜欢
最新推荐
热门推荐
更多推荐
【转帖】MSDS焦亚硫酸钠
2006/10/25
各位大佬有没有快速测锰的设备推荐?
原创
2024/06/03
【讨论】怎样鉴别彭化食品袋子里装的气体?
2008/09/23
【求助】问个白痴的问题
2007/08/17
【原创】滴定液的有关知识
2006/09/02
锰铁怎么溶解?
求助
2015/01/21
【求助】请问这个试剂的纯度是哪种级别的?
2011/04/04
天平为什么称不准?4
分享
2022/06/25
短链氯化石蜡
原创
2024/10/30
阴性的对照
求助
2024/10/30
100mmol/L对苯醌乙腈溶液的颜色
已应助
2024/10/29
硬度滴定空白是否可以写0mL
已应助
2024/10/29
硝酸锌潮解如何处理
已应助
2024/10/26
二甲苯溶剂中有四种物质
原创
2024/10/24
分析仪器信号去噪方法
第十七届原创
2024/10/23
甜蜜素忘记震荡就衍生怎么办
原创
2024/10/23
气相色谱法同时测定实验废水中24种防腐剂含量
第十七届原创
2024/10/27
【仪器检测】石墨炉原子吸收法测定酱油中的铅
第十七届原创
2024/10/28
表层水温表能不能把金属外壳去掉后,按照玻璃液体温度计进行校准?出具带CNAS logo证书?
已应助
2024/10/26
又遇气相色谱GC-2014新出问题,求大神们指导解决
求助
2024/10/28
记录的修改是不是必须写名字全称,能用名字的首字母代替吗?
讨论
2024/10/30
求助!GB23200.121的液质方法
求助
2024/10/30
天美GC7900严重拖尾
求助
2024/10/31
报告变更需要在新的报告中写明更改内容吗?
已应助
2024/10/28
应该怎么校准电导池?本视频告诉您
第十五届原创
2022/11/30
质谱仪五大组成结构及作用
分享
2022/11/30
飞行时间质谱仪的工作原理简介
分享
2022/11/30
GC/MS-O分析银杏外果皮气味成分
第十五届原创
2022/11/30
质谱仪--质谱仪原理介绍和原理公式
分享
2022/11/30
质谱仪常用的性能指标有哪些
分享
2022/11/30
质谱仪的应用介绍
分享
2022/11/30
实验室分析仪器--有机质谱仪器的性能指标
分享
2022/11/30
品牌合作伙伴
岛津
日立科学仪器
珀金埃尔默仪器(上海)有限公司(PerkinElmer)
日本电子株式会社
丹纳赫
安捷伦
赛默飞世尔科技
普析通用
欧波同
天美
天瑞仪器
德国耶拿
海能技术
马尔文帕纳科
磐诺科技
上海仪电科仪
梅特勒托利多
聚光科技
莱伯泰科
盛瀚
多宁生物
丹东百特
科哲
卓立汉光
屹尧科技
华谱科仪
宝德仪器
优莱博
HORIBA
布鲁克核磁
举报帖子
执行举报
点赞用户
好友列表
加载中...
正在为您切换请稍后...