摘要:电化学合成尿素法是一种相对绿色环保的尿素制备方法,其反应产物中,除目标产物尿素外,还含有大量NO3-、NO2-、CO32-等杂质离子,这些离子易对尿素测定产生干扰。本文基于多孔石墨碳柱特殊的极性保留效应,并结合其柱流失远小于普通反相C18色谱柱,在极低紫外吸收波长下基线稳定好的优势,利用多孔石墨碳柱建立了电合成尿素产物中尿素的高效液相色谱分析方法。经过一系列相关色谱条件优化后,选择HypercarbTM多孔石墨碳柱(100 mmí4.6mm,5 μm),采用水-25 mmol/L甲基磺酸溶液为流动相进行梯度洗脱分离,柱温为30 ℃,流速为1.0 mL/min,进样量为25.0 μL,检测波长190nm,可实现不受电解液中其它杂质如NO3-、NO2-、CO32-等离子干扰的尿素测定。结果表明,单次样品分析可在15 min内完成,尿素在0.1~100mg/L质量浓度范围内线性关系良好,相关系数(r2)大于0.9988;方法的检出限(S/N=3)和定量限(S/N=10)分别为0.028mg/L、0.093 mg/L,相对标准偏差(RSD)小于0.79%,低、中、高三个浓度下的加标回收率在112.0%~118.4%之间。最后对所接收的实际电解液样品进样分析,结果显示尿素色谱峰峰形良好,且未受到其他杂质峰的干扰。该方法前处理简单,方便快捷,尿素与其它相关离子分离完全,不受干扰,结果准确可靠,特异性好,可用于实际电合成尿素电解液产物中微量尿素及其它相关离子的检测。
关键词:多孔石墨碳柱;高效液相色谱;尿素;阴离子
前言
尿素(Urea),又称脲或碳酰胺,是最简单的有机化合物之一,其化学性质并不稳定,在酸、碱或酶的作用下易水解,在高温下发生缩合反应。尿素广泛存在于自然界中,是哺乳动物和两栖动物氮排泄的主要代谢产物,还可作为农作物肥料、动物饲料、日用品、炸药等产品的原料;也是许多日常护肤产品的主要成分,可用于临床中各种皮肤病的治疗[1],与日常生活息息相关。
电化学合成尿素法可以在一定程度上弥补传统尿素制备所带来的的能源消耗和环境污染问题[2]。该方法主要是以N2或其它氮源和CO2作为原料,在水中电耦合生成尿素[3]。由于亚硝酸盐的解离能更低,在合成反应中有利于节约成本、降低能耗[4],可采用碳酸氢根和硝酸盐或亚硝酸盐电解液与CO2一同反应进行制备[5]。在电化学合成法反应液中,鉴于方法产率较低,除含有少量产物尿素外,还存在大量NO3-、NO2-、CO32-等杂质离子,易对尿素测定产生干扰。
目前国内外关于尿素,特别是微量及痕量尿素的检测方法,已有多项报道,主要有脲酶法[6-8]、比色法[9, 10]、红外光谱法[11]及高效液相色谱法[12, 13]等等。刘灵辉等[7]采用脲酶-谷氨酸脱氢酶偶联法,改进后可实现游泳池水中尿素含量的自动化分析;Chen等[9]和付智慧[10]分别选取了二乙酰一肟和对二甲氨基苯甲醛为显色剂,使其与尿素发生显色反应后,利用分光光度计进行测定;谭良锋等[14]采用中红外光谱法进行车用尿素的定量分析。其中,脲酶法和比色法需要对样品进行复杂的预处理操作,灵敏度和精确度有所损失;红外光谱法一般可直接进样,方便快捷,但灵敏度相对较低;高效液相色谱具有检测快速、便捷灵敏等优点,由于尿素属于强极性化合物,难以在传统液相色谱柱上显著保留,常常采用亲水作用色谱柱[15]、柱前衍生-荧光检测法[12, 16]、串联质谱法[17]等来提高检测灵敏度。其中,Zhang[12]等人采用荧光检测器对酒精饮料中尿素的进行测定,检出限为0.003 mg/L;Kramer M[17]等人对比了串联质谱和荧光检测两种色谱方法检测饲料中的尿素含量,检出限分别为3 mg/kg和2 mg/kg。

1 实验部分
1.1 仪器、试剂与材料
U3000高效液相色谱仪(美国Thermo公司);;HypercarbTM多孔石墨碳柱,100 mmí4.6 mm,(美国Thermo公司);AL204 电子天平(梅特勒-托利多公司);Millipore-Q A10超纯水机(美国Merck Millipore公司);100~1000 μL移液枪(北京大龙公司);100mL容量瓶。
硫酸(96%,国药集团化学试剂有限公司);磷酸(85%水溶液)、甲基磺酸(MSA,99.5%)、尿素(99.5%,生物分子级)、无水碳酸钠(优级纯)、无水碳酸氢钠(优级纯)、氢氧化钠(优级纯)均为上海阿拉丁生化科技公司;水中NO3-、NO2-、NH4+标准溶液,100 mg/L,上海市计量测试技术研究院。
1.2溶液配制
1.2.1标准溶液的配制
尿素标准溶液:准确称量100.5 mg尿素至100 mL容量瓶中并用超纯水定容,得到1000 mg/L的尿素标准溶液,备用;根据情况分别取适量标准储备液,用超纯水稀释成不同浓度的标准溶液。
模拟电解液溶液:准确称取1.011 g硝酸钾和1.060 g碳酸钠至100 mL容量瓶中用超纯水定容,得到0.1 mol/L电解液,备用。
模拟电解液尿素样品溶液:准确称取1.011 g硝酸钾、1.060 g碳酸钠和100.5 mg尿素至100 mL容量瓶中并用超纯水定容,再利用0.1 mol/L电解液逐级稀释得到0.1 mol/L电解液背景下的系列浓度尿素样品溶液。
1.2.2样品前处理方法
电合成尿素的实际样品溶液以0.22 μm的滤膜过滤,即得待测样品溶液。
1.3色谱条件
色谱柱:HypercarbTM多孔石墨碳柱(100 mm í4.6 mm,5 μm); 柱温:30 ℃;流动相:
A:H2O;B:25 mmol/L MSA;流速:1 mL/min;进样量:25.0 μL;检测器:二极管阵列检测器,190 nm;梯度洗脱程序:0-2 min,2% B;2-8 min,40% B;8-15 min,2% B。
2 结果与讨论
2.1色谱条件的优化
2.1.1色谱柱和检测波长的选择
参考文献[16,22]色谱条件利用C18柱对尿素进行测定,但在实际分析中发现,高浓度的NO3-与尿素难以分离,干扰现象严重,如图1所示。因此我们尝试选择Hypercarb柱,发现NO3-会受到PGC较强的保留作用,使得NO3-出峰延后,远离尿素峰。这可能是由于NO3-所带孤对电子与PGC表面产生电子间相互作用,在酸性条件下,阴离子的保留性增强,而在碱性条件下反之。

图 1 C18柱下的尿素、硝酸钾及混合标样的色谱图
在对比了尿素以及其他相关杂质离子的紫外光谱图后发现,尿素紫外吸收小,而待测样品中尿素浓度很低,在190 nm这一极端波长下,尿素才有较大的响应,满足灵敏度的要求,而在195 nm或更高波长下,难以检测到微量尿素,灵敏度有所欠缺。同时,在190 nm低波长的极端检测条件下,PGC柱的背景噪音相较于C18柱等传统色谱柱而言更小,可在尿素检测时减少背景干扰。综合考虑选择190 nm作为检测波长,而在通常情况下,小于195 nm是无法进行紫外检测的。
2.1.2流动相的选择
鉴于尿素在碱性环境下不稳定且易分解,需在中性或酸性流动相中进行分离。选择磷酸、硫酸以及甲基磺酸三种常见酸性流动相,在相同色谱条件下,分别考察10 mmol/L磷酸、硫酸和5 mmol/L甲基磺酸下的分离效果,见图2。结果表明,磷酸洗脱得到的尿素峰值响应较低,且噪音较大,影响检测灵敏度;而以硫酸为流动相时,在尿素保留时间附近出现倒峰,干扰尿素测定。采用甲基磺酸溶液进行淋洗时,可得到基线平稳、峰形优良的尿素液相色谱图。故最终选择甲基磺酸溶液作为流动相。
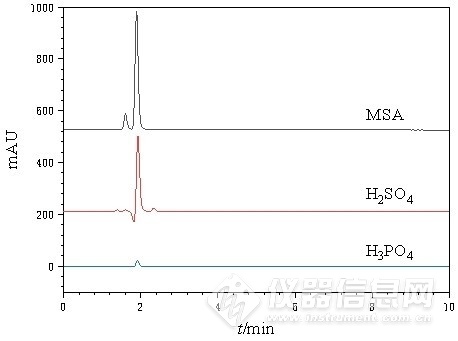
图 2 三种酸性流动相下的尿素标样色谱图
2.1.3流速和柱温的优化
流动相的流速以及色谱柱的柱温均会对待测物的出峰时间以及峰宽造成影响,流速的提高使得柱压增大,从而导致出峰提前;而柱温的改变也会影响待测物与固定相之间的相互作用,发生出峰提前或延后的现象。为确立最佳流速及柱温,在PGC柱上,以5.0 mmol/L甲基磺酸作为淋洗液,对所配制的50 mg/L尿素标样进样,首先考察了0.3~1.2 mL/min不同流速对分离效果的影响,结果表明,随着流速增加,尿素保留时间缩短,且峰宽变窄;当流速为1.0 mL/min时,有效塔板数较大,分离效果好。其次考察了20~40 ℃不同柱温对检测结果的影响,数据显示,柱温升高会导致保留时间略有前移,30 ℃下的有效塔板数最高。综合考虑保留时间、塔板数以及分析效率、峰形等因素,最终确定流速为1.0 mL/min,柱温为30 ℃。
表 1 不同流速下尿素标样的色谱数据
| Flow rate/(mL/min) | tR/min | Noise/mAU | Neff |
| 0.3 | 6.167 | 0.061 | 2643 |
| 0.4 | 4.643 | 0.043 | 2487 |
| 0.5 | 3.727 | 0.071 | 2678 |
| 0.6 | 3.109 | 0.048 | 2441 |
| 0.7 | 2.673 | 0.072 | 2167 |
| 0.8 | 2.350 | 0.043 | 2131 |
| 0.9 | 2.095 | 0.070 | 2140 |
| 1.0 | 1.890 | 0.063 | 2471 |
| 1.1 | 1.717 | 0.062 | 2448 |
| 1.2 | 1.580 | 0.043 | 2048 |
表 2 不同柱温下尿素标样的色谱数据
| Temperature/℃ | tR/min | Noise/mAU | Neff |
| 20 | 1.963 | 0.072 | 2375 |
| 25 | 1.921 | 0.051 | 2328 |
| 30 | 1.888 | 0.070 | 2381 |
| 35 | 1.854 | 0.043 | 2192 |
| 40 | 1.826 | 0.063 | 2145 |
2.1.4浓度优化及干扰离子实验
在流动相淋洗浓度优化实验中,由于样品中除尿素外还含有大量NO3-、NO2-、NH4+、HCOO-、HCO3-等杂质离子,在选择流动相浓度时需考虑尿素与杂质的分离度。本研究初步尝试等度淋洗的方法。分别选取了10 mmol/L、5 mmol/L、2.5 mmol/L三个浓度的甲基磺酸溶液作为淋洗液进行洗脱。检测结果表明,当淋洗液浓度不断降低时,尿素保留时间未有明显变化,而NO3-等离子的保留时间发生后移。当甲基磺酸浓度为2.5 mmol/L时,NO3-的出峰时间延长至20 min,拉长了检测时间,影响检测效率。而以5 mmol/L甲基磺酸溶液洗脱时,NO2-和尿素的色谱峰产生部分重叠(图2.a),影响尿素分析的准确性。
鉴于此,为实现尿素与其他离子的完全分离,提高分析效率,进一步开发梯度洗脱的方法。最终确定洗脱程序见1.3。在此梯度淋洗条件下,对尿素混合杂质样品及杂质混合样品分别进样分析。结果表明,尿素的检测未受到NO2-等其他杂质离子的干扰(图2.b),峰形窄而尖,有利于定性定量分析,且分析时间仅15 min,提高了检测效率。

图 3 尿素杂质混合样品(上)及杂质混合样品(下)色谱图 a等度洗脱b梯度洗脱
a. isocratic eluting; b.gradient eluting
Peak identifications:1. urea;2.NO2-;3. NH4+;4. NO3-.
2.2方法学考察
2.2.1线性关系、检出限与定量限
根据1.3所述色谱条件进行测定。待仪器稳定后,对配制好的0.1、1.0、5.0、10、25、50、100 mg/L一系列电解液尿素线性标准溶液,浓度从低到高依次进样分析,每个浓度重复测定3次。分别以峰面积和峰高的平均值为纵坐标,尿素质量浓度为横坐标绘制标准曲线,可得到对应的线性回归方程。结果表明,峰面积和峰高与浓度均呈现良好的线性关系,相关系数r2分别为0.9997和0.9988。同时取噪音0.098 mAU,以S/N=3计算方法的检出限为0.028 mg/L,以S/N=10计算方法的定量限为0.093 mg/L。
2.2.2重复性
为考察方法的重复性,同样在最佳色谱条件下,对配制好的10 mg/L、50 mg/L、100 mg/L三个浓度的标准溶液样品分别进行连续进样分析,每个浓度平行测定8次,记录相应的峰面积和峰高。结果见表3,保留时间及峰面积、峰高的相对标准偏差(RSD)在0.065%~0.79%之间,说明该方法的检测重复性良好。
表 3 三种浓度下尿素标样的精密度(n=8)
| No. | 10 mg/L | 50 mg/L | 100 mg/L |
| tR/ min | Peak area/ (mAU*min) | Peak height/ mAU | tR/ min | Peak area/ (mAU*min) | Peak height/ mAU | tR/ min | Peak area/ (mAU*min) | Peak height/ mAU |
| RSD(%) | 0.17 | 0.27 | 0.79 | 0.081 | 0.065 | 0.64 | 0.072 | 0.084 | 0.67 |
2.2.3加标回收率
通过对实际电解液样品进行加标回收实验,以考察方法的准确度。选取一个预处理后的0.1 mol/L电解液体系下实际电合成尿素的样品溶液为基底,加入电解液标准样品,得到位于线性范围内的低、中、高三个浓度待测样品,平行测定6次,根据结果计算回收率和RSD值,具体见表4。所得加标回收率在112.0%~118.4%之间,RSD在3.1%~4.6%之间,表明该方法准确度良好,能满足实际样品基本检测需求。
表 4 电解液中尿素的加标回收率和相对标准偏差(n=6)
| Compound | Background/(mg/L) | Added/(mg/L) | Detected/(mg/L) | Recovery/% | RSD/% |
| Urea | 0.126 | 0.080 | 0.221 | 118.2 | 4.6 |
| 0.10 | 0.238 | 112.0 | 3.1 |
| 0.12 | 0.268 | 118.4 | 4.6 |
2.3实际样品检测
采用该创新性的分析体系,对所接收的0.1 mol/L硝酸钾和碳酸钾电解液体系下进行电合成尿素的多个实际样品进行检测。由色谱图分析,在1.5 min左右出现一较高色谱峰,在对照了干扰离子实验的结果后难以对其进行归属,猜测其为在实际电解合成尿素过程中产生的其他杂质。同时NO2-以及高浓度的NO3-等杂质离子的保留时间与标样存在差异,可能是实际样品中的其他未知化合物对其在PGC柱上的保留造成影响,但并未对低浓度的尿素测定造成干扰,并利用峰面积进行定量计算得该样品中尿素浓度为0.126 mg/L,其余样品中尿素浓度分别为0.0963 mg/L、0.148 mg/L、0.152 mg/L。说明将该方法应用于实际电合成生产尿素的产物检测是可行的。
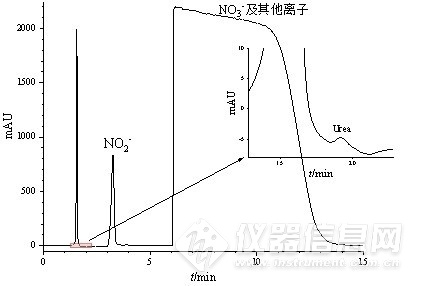
图4 电合成尿素实际样品色谱图
2 结论
本研究建立了将PGC柱应用于高效液相色谱来测定电解液体系下微量尿素的分析方法。针对电合成尿素样品的特点,选择Hypercarb柱,其特殊的保留效应能将尿素与其它高浓度干扰离子轻易分开,避免了高浓度基体的干扰,在190 nm的超低检测波长下,既提高尿素的灵敏度,并保证较低的背景噪音和基线的稳定性。该方法灵敏度高,准确性好,且前处理步骤简单,检测效率高,为电合成尿素的测定提供了一种新的分析手段。
参考文献:
[1] Dirschka T. Int J Clin Pract, 2020, 74:3
[2] Zhu X, Zhou X, Jing Y,et al. Nat Commun, 2021, 12(1): 4080
[3] Chen C, Zhu X, Wen X,et al. Nat Chem, 2020, 12(8): 717
[4] Meng N N, Huang Y M,Liu Y, et al. Cell Rep Phys Sci, 2021, 2(12): 100378
[5] Feng Y G, Yang H,Zhang Y, et al. Nano Letters, 2020, 20(11): 8282
[6] GB/T 29661-2013. Beijing: General Administration of QualitySupervision, Inspection and Qurantine of the People's Republic of China, 2013
GB/T 29661-2013. 北京:中华人民共和国国家质量监督检验检疫总局, 2013
[7] Liu L H, Gu S Y, Luo QY, et al. Occupation and Health, 2018, 34(18): 2564
刘灵辉, 谷素英, 骆秋云,等. 职业与健康,2018,34(18): 2564
[8] Ali S M U, Ibupoto ZH, Salman S, et al. Sensor & Actuat B-Chem, 2011, 160(1): 637
[9] Chen L, Ma J, Huang Y,et al. Limnol Oceanogr Meth, 2015, 13(6): 303
[10] Fu Z H. YunnanChemical Technology, 2021, 48(7): 99
付智慧. 云南化工, 2021, 48(7): 99
[11] Bai J, Wang H. Foodand Fermentation Industries, 2020, 46(8): 267
白静,王会. 食品与发酵工业, 2020, 46(8): 267
[12] Zhang J, Liu G X,Zhang Y, et al. J Agric Food Chem, 2014, 62(13): 2797
[13] Wang R F, Xu T, Wei F,et al. Chinese Journal of Veterinary Drug, 2020, 54(2): 39
王瑞菲,徐婷,魏凤,等. 中国兽药杂志, 2020, 54(2): 39
[14] Tan L F, Chu G Y, Huang J Y. Modern Chemical Research, 2021 (14): 30
谭良锋,邹国雁,黄俊源. 当代化工研究, 2021 (14): 30
[15] Bai D W, Cheng D, Zhao H. Strait Pharmaceutical Journal, 2020, 32(6):62
柏大为,程冬,赵慧. 海峡药学, 2020, 32(6): 62
[16] Zeng Q, Zhang J, Xu D M, et al. Chinese Journal of Chromatography,2015, 33(1): 80
曾琪, 张缙, 徐敦明,等. 色谱, 2015, 33(1): 80
[17] Kramer M, Fry H,Kappenstein O. Food Addit Contam Part A-Chem, 2021, 38(6): 931
[18] Pereira L. J Liq Chromatogr Relat Technol, 2008, 31(11-12): 1687
[19] Grinevich O, Khesina Z, Buryak A. Rev Anal Chem, 2021, 41(1): 1
[20] Jin C H, Eom H Y, Bae S J, et al. J Anal Sci Technol, 2021, 12(1): 26
[21] Yokoyama T, Andoh Y, Kunisawa T, et al. Anal Sci, 2023, 39(3): 285
[22] Liu Y, Wang Y, Huang Y M, et al. Chinese Journal of AnalysisLaboratory, 2021, 40(10): 1216
刘洋, 王意, 黄艳梅,等. 分析试验室, 2021, 40(10): 1216



































