2005yfc
第4楼2009/06/21
该仪器常用电灯电流为10mA流,负高压正常使用范围为40-80。我也试验过灯电流大小对吸光度的影响试验,为6VmA时,负高压47,20ug/L标准吸光度为0.0462,变化不大。
方法为:PE800,灯电流10mA,狭缝0.7L,负高压63,自动进样20ul,铅标准购自国家标准物质中心。工作曲线法,标准系列:10、20、40ug/L,每个标准点均加入了0.10ml低浓度的质控样,标准线性0.999以上;样品测定:0.10ml样不消化直接加入0.90ml稀释剂中混匀测定。各测定液中含改进剂0.20ml【1.66g/L二氯化钯-2.5%硝酸-2.5%T-X100】。干燥:110/10-30,130/5-25,灰化:850/10-25,原子化:1850/0-4,清除:2450/1-3.
quexianyin
第6楼2009/06/21
我有几点看法如下,不当之处,请大家指教!
PE的我没用过,是不是塞曼扣背景,如果是,那不存在着背景扣过头情况!往往是氘灯扣背景背景吸光度超过0.5就扣不准,所以氘灯扣背景的石墨炉,当你用基改时候,我认为这个石墨炉是个摆设!做不出准确数字的!
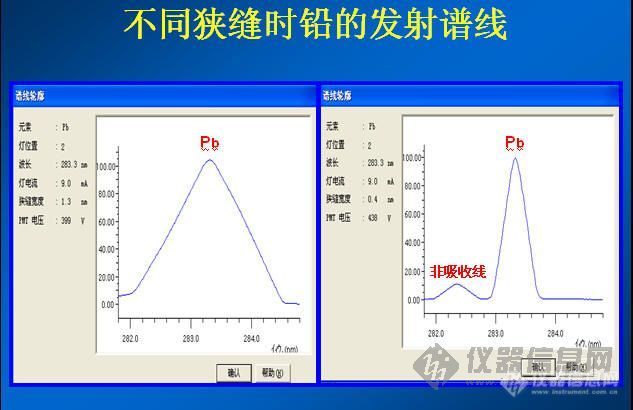
关于楼上说的谱线干扰,我觉得可能性比较小!石墨炉,一般都以峰高测定的!
发帖的用什么钯盐,我是觉得不太好,如果你的仪器是塞曼扣背景,您可以用磷酸二氢铵,浓度为50 g/L,升温程序改掉!不要用厂家推荐的!当然各个仪器红外测温有点偏差,小镜子或许有点污垢!你程序升温可以试试改成如下:
95/10-30,130/5-25,灰化:600/10-25,原子化:1850/0-3,清除:2500/0-3.
另:加基体改进剂的,一般都建议你用峰高测定!你40ug/ml的点吸光度才这么点,我20这个点,用217的分析线都有0.3左右!你是吸光度太小了!虽然283这条线灵敏度会下降50%左右,但是也不至于这么点吸光度!另外,你石墨管装上去以后,有没有认真调试过光路,让其通过石墨管最中间,而且进样针为止要调试到合适的位置??如果这两个方面调试的不对,出现这类低级失误,那咱们探讨你这原因就感觉白忙呼一场,呵呵!
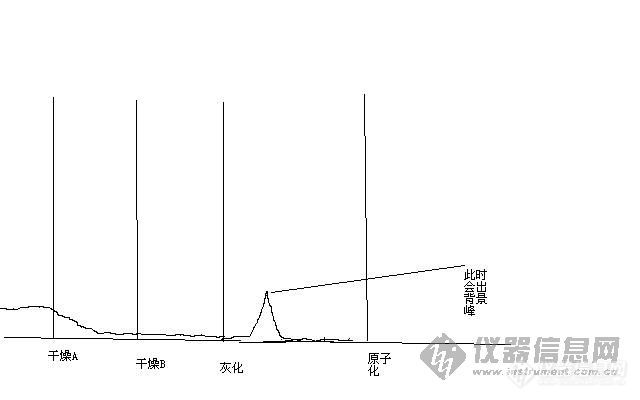
你试试你原来设置的升温是否有跑的可能性,你标准曲线可以加几个点,比如:0、5、10、20、30、40 看是否是凹形的!凹形说明灰化温度太高!一般原子化0.7秒左右开始出峰,出峰时间太早,是灰化时间太长了!我画了个草图如附件!关于原子化温度太高还是太低了,你自己多去试试钯!看看吸光度变化多少!你要让我形象的说,我还真说不出来,要是我做,我还能看的出来!
石墨炉这玩意没有火焰的知识经典,理论也未完善成熟!!而且不直观,所以这个东西一定要做的人,才能试两下才能找的到原因!找原因试条件的时候,低级失误不要出现,呵呵,否则试的就是没有论证性了!凡事得多开动脑筋想想吧,这样会有受益的!