
离子交换型的固相萃取柱,必须考虑离子交换的容量。不同厂家的小柱离子交换容量稍有差异。下表附SPE小柱的容量和洗脱参数
SPE柱上样容量和洗脱体积的选择
规格 | 最大上样量 | 最小洗脱体积 |
100mg/1mL | 5mg | 250µL |
200mg/3mL | 10mg | 500µL |
500mg/6mL | 25mg | 1.2mL |
1g/6mL | 50mg | 2.4mL |
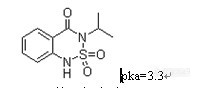
添加浓度 (μg/L) | 回收浓度 (μg/L) | 平均回收值 (μg/L) | 平均回收率 (%) | 相对标准偏差 (%) |
1 | 0.75 | 0.72 | 72.40 | 5.93 |
0.67 | ||||
0.72 | ||||
0.70 | ||||
0.78 | ||||
2 | 1.62 | 1.63 | 81.30 | 1.23 |
1.66 | ||||
1.60 | ||||
1.61 | ||||
1.64 | ||||
5 | 4.02 | 4.24 | 84.80 | 4.16 |
4.10 | ||||
4.27 | ||||
4.38 | ||||
4.43 | ||||
10 | 8.24 | 8.45 | 84.45 | 2.81 |
8.35 | ||||
8.77 | ||||
8.62 | ||||
8.25 | ||||
100 | 90.24 | 9.12 | 91.15 | 2.86 |
87.15 | ||||
91.77 | ||||
92.62 | ||||
93.95 |
批间 | 添加浓度(μg/L) | |||||||||
1 | 2 | 5 | 10 | 100 | ||||||
平均 回收率% | RSD% | 平均 回收率% | RSD% | 平均 回收率% | RSD% | 平均 回收率% | RSD% | 平均 回收率% | RSD% | |
1 | 72.40 | 5.93 | 81.30 | 3.49 | 84.80 | 6.16 | 84.45 | 3.59 | 91.15 | 2.86 |
2 | 75.37 | 6.12 | 80.47 | 5.37 | 84.74 | 7.55 | 87.46 | 4.68 | 90.05 | 3.86 |
3 | 70.09 | 7.85 | 80.80 | 6.57 | 83.10 | 8.17 | 83.21 | 5.39 | 89.53 | 4.16 |
4 | 76.73 | 4.90 | 78.50 | 8.35 | 82.90 | 5.11 | 85.95 | 5.72 | 88.27 | 5.93 |
平均值 | 73.65 | 6.20 | 80.25 | 5.95 | 83.88 | 6.75 | 85.27 | 4.84 | 89.75 | 4.20 |
RSD% | 12.95 | 10.79 | 9.43 | 7.00 | 5.75 | |||||
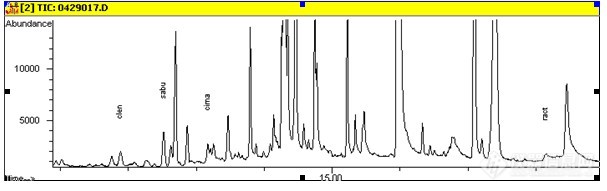
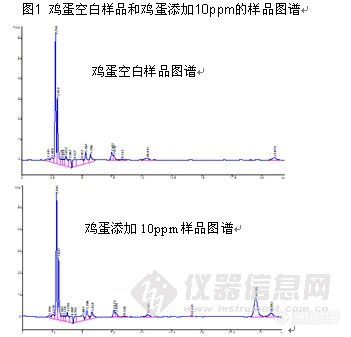
表1 保留时间与峰面积的稳定性数据
浓度mg/mL | 指标 | 1# | 2# | 3# | 4# | 5# | 6# | 平均值 | RSD% |
1.0 | 保留时间(min) | 18.830 | 18.829 | 18.829 | 18.838 | 18.840 | 18.834 | 18.833 | 0.026 |
峰面积 | 89 | 81 | 84 | 88 | 84 | 80 | 84 | 4.286 | |
5.0 | 保留时间(min) | 18.949 | 18.952 | 18.947 | 18.949 | 18.950 | 18.946 | 18.949 | 0.011 |
峰面积 | 423 | 440 | 438 | 439 | 437 | 438 | 436 | 1.461 |
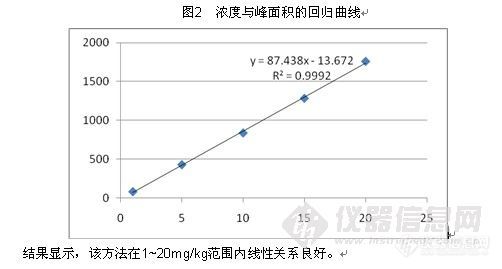
表2 标准校正曲线实验数据
浓度 mg/kg | 峰面积 | 峰面积 | 峰面积 均值 |
1.0 | 89 | 79 | 84 |
5.0 | 423 | 440 | 431 |
10.0 | 832 | 844 | 838 |
15.0 | 1265 | 1299 | 1282 |
20.0 | 1689 | 1823 | 1756 |
添加浓度(mg/kg) | 峰面积 | 计算含量 | 回收率(%) |
1.0 | 19.9 | 1.158892 | 115.89 |
1.0 | 21.0 | 1.214532 | 121.45 |
2.0 | 41.7 | 2.261587 | 113.08 |
2.0 | 40.8 | 2.216062 | 110.80 |
10.0 | 188.8 | 9.702247 | 97.02 |
10.0 | 219.6 | 11.26018 | 112.60 |
核桃
第1楼2010/09/15
2、环境检测应用
1)水中酚类检测(Cleanert PEP, P/N: PE0603)
1 实验材料:
1.1 SPE小柱:Cleanert PEP 500mg/6mL
1.2 7种酚类:苯酚,4-硝基酚,间甲酚,2-氯酚,2,4-二氯酚,2,4,6-三氯酚,五氯酚
2 实验过程
2.1.样品前处理方法
1) 活化:依次用5mL甲基叔丁基醚(10:90,V/V)、5mL甲醇活化富集柱、5mL去离子水活化Cleanert PEP柱,5mL/min
2) 上样:1L的水样过柱,<5mL/min
3) 淋洗:10mL去离子水淋洗小柱,5mL/min,然后真空抽干20min
4) 洗脱:2mL甲醇/甲基叔丁基醚(10:90,V/V)分两步洗脱,收集洗脱液至尖嘴瓶中
5) 浓缩:将收集的2mL洗脱液,氮气浓缩至1mL
2.2.色谱条件
色谱柱:Venusil MP C18(4.6*150,5µm)
流动相:A:1%乙酸
B:1%乙酸甲醇
检测器:UV检测器
梯度洗脱程序:
时间 | 流动相比例 | 流速(mL/min) | 检测波长(nm) |
0-15min | A:B=50:50 | 1 | 275 |
15-30min | A:B=15:85 | 1.8 | 295 |
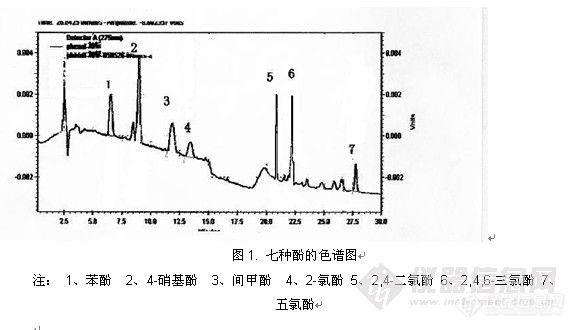
表1. 七种酚类在水中的添加回收率结果
| 加标 | 平均值 | 标准偏差 | 平均回收率(%) | ||
1 | 2 | 3 | ||||
苯酚 | 1.367 | 1.541 | 1.524 | 1.477 | 0.096 | 100.3 |
4-硝基酚 | 1.229 | 1.308 | 1.430 | 1.322 | 0.101 | 90.0 |
间甲酚 | 1.294 | 1.540 | 1.548 | 1.461 | 0.144 | 106.3 |
2-氯酚 | 0.527 | 0.684 | 0.641 | 0.617 | 0.081 | 100.6 |
2,4-二氯酚 | 1.305 | 1.613 | 1.621 | 1.513 | 0.180 | 92.8 |
2,4,6-三氯酚 | 1.365 | 1.609 | 1.511 | 1.495 | 0.123 | 90.3 |
五氯酚 | 1.259 | 1.487 | 1.472 | 1.406 | 0.128 | 95.6 |
核桃
第2楼2010/09/15
2)水中的多环芳烃固相萃取方法(Cleanert PEP, P/N: PE0603)
1. 材料
1.1 目标物成分
萘,苊,苊烯,芴,菲,蒽,荧蒽,芘,屈,苯并(a)蒽,苯并(k)荧蒽,苯并(a,h)荧蒽,苯并(a)芘,苯并(g,h,i)芘,茚并(1,2,3-cd)芘
1.2.SPE柱:Cleanert PEP(60g/3mL)
2. 样品处理:1L 水中加入20mL10%的硝酸
3. SPE方法
1) 活化:5mL异丙醇和5mL水分别依次活化PEP柱
2)上样:把处理好的水样过PEP柱
3) 淋洗:用5mL淋洗液(300mL水+700mL甲醇+2.1 g Na2HPO4+2.04 g KH2PO4)淋洗
4)抽干:抽真空干燥柱管30min
5)洗脱:用4mL洗脱液(90mL异丙醇+10mL冰醋酸+200mL甲苯,加入到1L石油醚中),混合均匀洗脱,收集洗脱液
6)浓缩,定容
4. 色谱条件:
色谱柱:Venusil PAH专用柱(4.6×250mm,5μm,200Å)
样 品:溶于MeOH:CH2Cl2(1:1)16PAHs标品, 用MeOH:CH2Cl2(1:1)稀释10倍
流速:1.2mL/min
进样量:10μL
柱 温:30℃
波 长:254nm
甲醇/水 线性梯度洗脱,梯度表如下
T(min) | MeOH(%) | H2O(%) |
0 | 85 | 15 |
2 | 85 | 15 |
7 | 95 | 5 |
40 | 95 | 5 |

核桃
第3楼2010/09/15
3、药物分析中的应用
1)血浆中油酸及其代谢物LC-MS分析中的应用(Cleanert PAX, P/N: AX0603)
图片如下:
2.提取及净化方法:见下图
3.检测条件:
仪器:API Qtrap 3200, 美国Applied Biosystem公司;LC-20A高效[url=https://insevent.instrument.com.cn/t/5p]液相色谱,日本岛津公司
质谱条件:电离子喷雾源;负离子模式检测;扫描方式为选择反应监测(MRM)方式
用于定量分析的离子反应分别为:m/z 281.2® m/z 281.2 (油酸);m/z 315.2 ® m/z 315.2(油酸代谢物);m/z 269.2® m/z 269.2 (内标C17)
流动相为:ACN: 3mmol/L ammoniµm acetate=85:15
4.实验结果
浓度 | 10ng/mL(n=3) | 100ng/mL(n=3) | 2500ng/mL(n=3) |
油酸回收率(%) | 77 | 87 | 91 |
RSD | 2.9 | 0.9 | 0.2 |
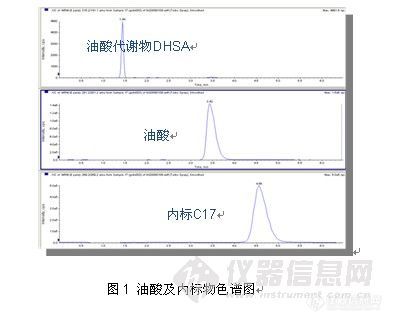
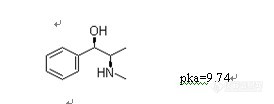
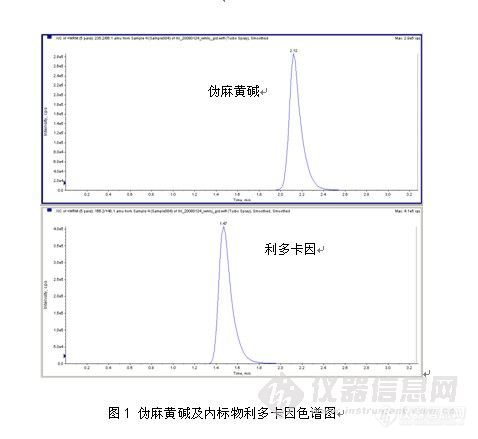
表1 血浆中伪麻黄碱回收率结果
浓度 | 10ng/mL(n=3) | 100ng/mL(n=3) | 2500ng/mL(n=3) |
伪麻黄碱回收率(%) | 77 | 79 | 85 |
RSD | 3.5 | 1.9 | 0.5 |
利多卡因回收率(%) | 88 | 92 | 87 |
RSD | 4.9 | 2.1 | 0.3 |