

〓猪哥哥〓
第1楼2010/10/02
一、仪器及试药
1. 药品与试剂
黄芪甲苷对照品(中国药品生物制品检定所,供含量测定用);醒脑再造丸;乙腈(色谱纯);其他均为分析纯。
2. 实验仪器
日本岛津LC-10AT VP高效液相色谱仪,ALLTECH ELSD2000型蒸发光散射检测器,SPD-10AT VP型自动进样器
3. 色谱条件
色谱柱:Phenomenex Gemini C18(200×4.6mm,5µm);流动相:乙腈- 水(36:64);流速:1.0ml/min;柱温:40℃;漂移管温度:105℃;载气流速:2.7L/min;Gain:8;理论板数按黄芪甲苷峰计算不低于4000。
4.样品分析方法
4.1对照品溶液的制备:精密称取黄芪甲苷对照品适量,加甲醇制成每1ml含0.1mg的溶液,即得。
4.2供试品溶液的制备 取重量差异项下的本品剪碎,取9g,精密称定,加硅藻土8g,研细,置索氏提取器中,加甲醇40ml,冷浸过夜,再加甲醇适量,加热回流6小时,提取液回收甲醇并浓缩至干,残渣加水20ml,微热使溶解,用水饱和的正丁醇振摇提取4次,每次40ml,合并正丁醇提取液,用氨试液充分洗涤2次,每次40ml,弃去氨液,正丁醇液蒸干,残渣加水10ml使溶解,放冷,通过D101 型大孔吸附树脂柱 (内径1.5cm,长12cm),以水50ml洗脱,弃去水液,再用40%乙醇30ml洗脱,弃去洗脱液,继用70%乙醇100ml洗脱,收集洗脱液,蒸干,用甲醇溶解并转移至5ml 量瓶中,加甲醇至刻度,摇匀,即得。
4.3测定方法:精密吸取对照品溶液10μl、20μl,供试品溶液20μl,注入液相色谱仪,测定,以外标两点法对数方程计算,即得。
〓猪哥哥〓
第2楼2010/10/02
二、方法与结果
1.提取方法的考察:
①取本品9g,剪碎,精密称定,加70%乙醇100ml,称定重量,加热回流4小时,放冷,称定重量,加乙醇补足减失重量,滤过,取续滤液25ml,蒸干,加水10ml使溶解,用水饱和正丁醇振摇提取4次,每次20ml,合并正丁醇液,用氨试液充分洗涤2次,每次40ml,弃去氨液,正丁醇液蒸干,用甲醇溶解并转移到5ml量瓶中,加甲醇稀释至刻度,进行测定。
②取本品9g,精密称定,加硅藻土8g,研细,加甲醇40ml,冷浸过夜,再加甲醇适量,加热回流6小时,提取液回收甲醇并浓缩至干,残渣加水20ml微热使溶解,用水饱和正丁醇振摇提取4次,每次40ml,合并正丁醇提取液,用氨试液充分洗涤2次,每次40ml,弃去氨液,正丁醇液蒸干,残渣加水10ml使溶解,放冷,通过通过D101 型大孔吸附树脂柱 (内径1.5cm,长12cm),以水50ml洗脱,弃去水液,再用40%乙醇30ml洗脱,弃去洗脱液,继用70%乙醇100ml洗脱,收集洗脱液,蒸干,用甲醇溶解并转移至5ml 量瓶中,加甲醇至刻度,摇匀,进行测定。
③取本品9g,精密称定,加硅藻土8g,研细,置索氏提取器中,加甲醇40ml,冷浸过夜,再加甲醇适量,加热回流6小时,提取液回收甲醇并浓缩至干,残渣加水20ml微热使溶解,用水饱和正丁醇振摇提取4次,每次40ml,合并正丁醇提取液,用氨试液充分洗涤2次,每次40ml,弃去氨液,正丁醇液蒸干,残渣加水10ml使溶解,放冷,通过通过D101 型大孔吸附树脂柱 (内径1.5cm,长12cm),以水50ml洗脱,弃去水液,再用40%乙醇30ml洗脱,弃去洗脱液,继用70%乙醇100ml洗脱,收集洗脱液,蒸干,用甲醇溶解并转移至5ml 量瓶中,加甲醇至刻度,摇匀,进行测定。
采用上述三种提取方法进行试验,结果见表1。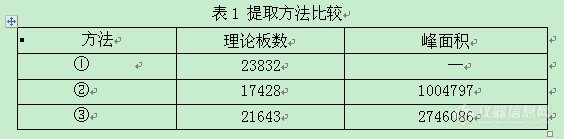
〓猪哥哥〓
第3楼2010/10/02
从上表得出,方法③的分离效果良好,通过D101大孔吸附树脂柱100ml洗脱液后无黄芪甲苷检出, 黄芪甲苷提取比较完全,因此我们采用正文所述的供试品溶液的制备方法。
2.色谱柱的选择及柱效的考察:
用十八烷基硅烷键合硅胶为填充剂;乙腈-水(36:64)为流动相;柱温40℃。
①采用Phenomenex Gemini C18(250×4.6mm,5µm)的色谱柱测定,黄芪甲苷对照品及供试品色谱图。
②采用PhenomenexR Luna C18(250×4.0mm,5µm)的色谱柱测定,黄芪甲苷对照品及供试品色谱图。
③采用DiamonsilTM M C18(250×4.0mm,5µm)的色谱柱测定,,黄芪甲苷对照品及供试品色谱图。
采用上述三种色谱柱进行试验,结果见表2。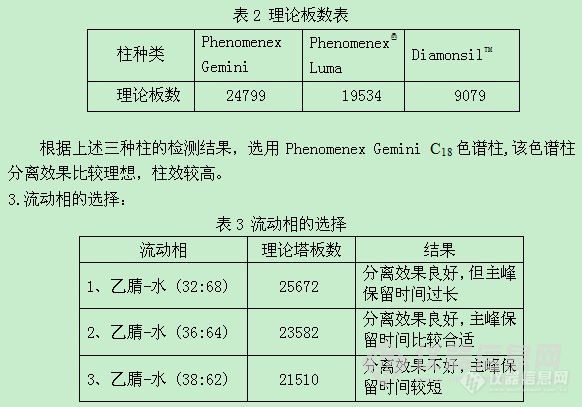
〓猪哥哥〓
第4楼2010/10/02
综合考察以上3个流动相比例,乙腈:水(36:64)分离效果良好,黄芪甲苷主峰保留时间适中,峰形好。
4.空白对照试验
为进一步考察试验的合理性,取不含黄芪的空白对照样品,依正文所述方法进行测定。结果显示,空白样品色谱在与黄芪甲苷峰相应的保留时间附近无干扰峰检出。
5.方法学考察
5.1标准曲线制备
精密称取黄芪甲苷对照品适量,加甲醇制成每1ml含0.1mg的溶液,分别精密吸取5、10、15、20、25µl测定,以对照品进样量(µg)的自然对数为横坐标,以峰面积的自然对数为纵坐标,绘制标准曲线,计算回归方程,Y=1.9526x+13.6286 (r=0.9997),结果表明黄芪甲苷在0.486-2.43µg范围内,呈线性关系,符合外标对数两点法定量测定的要求。
5.2精密度试验
按正文供试品溶液制备方法制备供试液,精密吸取供试液20µl,注入液相色谱仪,重复6次,测定其色谱峰面积值,RSD为0.92%。
5.3稳定性试验
取供试品溶液,每隔0,2,4,8,12,24h取样,按正文色谱条件测定。计算RSD为1.29%。结果表明24小时内供试品溶液中黄芪甲苷的含量基本稳定。
〓猪哥哥〓
第5楼2010/10/02
5.4回收率试验
精密称取已知含量的供试品(含量为 0.4352mg/丸)6份,分别精密加入对照品溶液(0.1067mg/ml)2.0 ml,依法测定,计算回收率,结果见表4。
表4 回收率试验结果
NO | 取样量(g) | 样品含量(mg) | 加标量(mg) | 测得量(mg) | 回收率(%) |
1 | 4.5393 | 0.2195 | 0.2134 | 0.4319 | 99.53 |
2 | 4.5434 | 0.2197 | 0.2134 | 0.4387 | 102.62 |
3 | 4.5430 | 0.2197 | 0.2134 | 0.4367 | 101.69 |
4 | 4.5455 | 0.2198 | 0.2134 | 0.4369 | 101.73 |
5 | 4.5393 | 0.2195 | 0.2134 | 0.4353 | 101.12 |
6 | 4.5390 | 0.2195 | 0.2134 | 0.4362 | 101.56 |
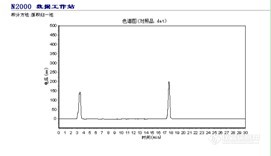
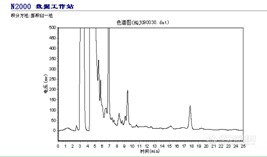
表5样品及黄芪甲苷的含量测定结果
厂家 | 含量(mg/丸) |
1 | 0.4352 |
2 | 0.4286 |
3 | 0.1799 |
4 | 0.4669 |
如表5所示,四批样品含量测定结果显示,根据《中国药典》2005年版一部黄芪的含量测定限度和该处方中黄芪药材的量,得出本品每丸含黄芪甲苷最低不得少于0.24mg,而上述测定结果比较切合实际有三批。因此,暂定本品每丸含黄芪甲苷(C41H68O14)计,不得少于0.24mg。
三、讨论
黄芪为方中君药,故采用测定黄芪甲苷的含量来控制其质量.关于黄芪甲苷的含量测定方法文献报道和载入标准的方法较多, 经薄层色谱法试验,黄芪甲苷与红参、三七中的某一皂苷成分薄层色谱行为一致,故无法采用薄层扫描法进行含量测定。黄芪甲苷只有紫外末端吸收,若采用UV法测含量,黄芪甲苷的浓度很大 难达到分离,而采用甲醇提取,水饱和正丁醇萃取,D101大孔吸附树脂进一步除杂后,再参照《中国药典》2005版一部黄芪项下的含量测定方法(HPLC-ELSD法)测定,该方法重现性好,准确度高。
参考文献
1.《中国药典》2005年版一部:212
2.辛颖,宋晓东,刘哲,张禅那,惠汝太.HPLC-ELSD法测定欣力胶囊中黄芪甲苷的含量.《药物分析杂志》2008,28(4):602
3.马淑杰.高效液相色谱-蒸发光散射法测定驴胶补血颗粒中黄芪甲苷的含量.时珍国医国药-2007:18(1)-98-99
4.王端统,林巨健,许玉丽等.HPLC-ELSD法测定脑清宁中黄芪甲苷的含量.中华实用医药杂志,2005,11(5):2

































