


〓猪哥哥〓
第8楼2010/10/29
庐山居士,你好
我觉得可能有几个原因:
1、色谱柱。因为最初使用的是盐酸为基质,对色谱柱可能有影响,更换一根色谱柱再试试。
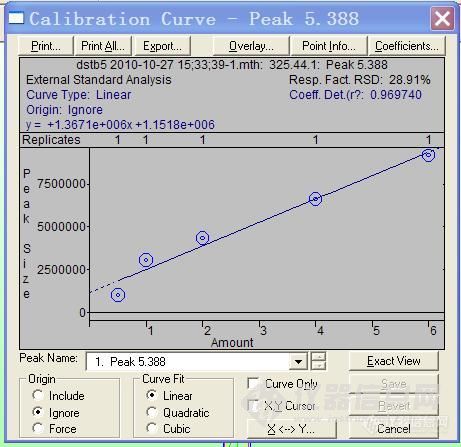
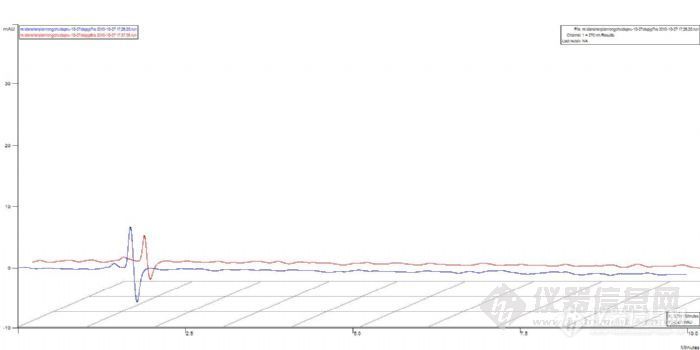
2、定量环。对于这个问题,更换一个六通阀或者直接更换一台仪器试试。不过这个可能性会比较小,因为原实验出现了溶剂峰。
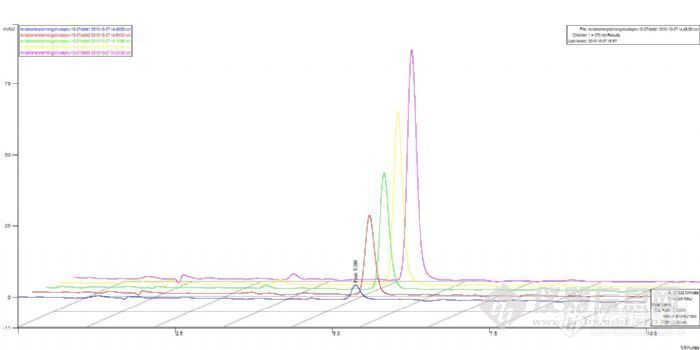
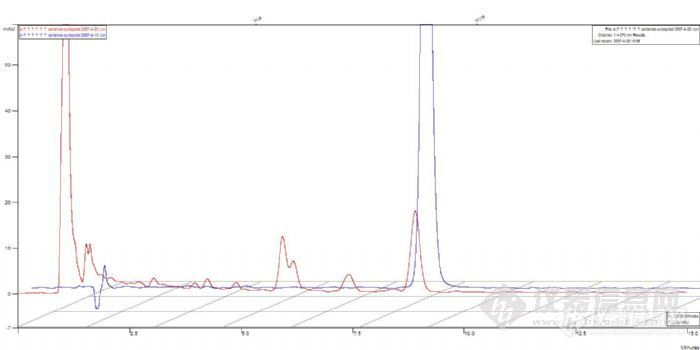
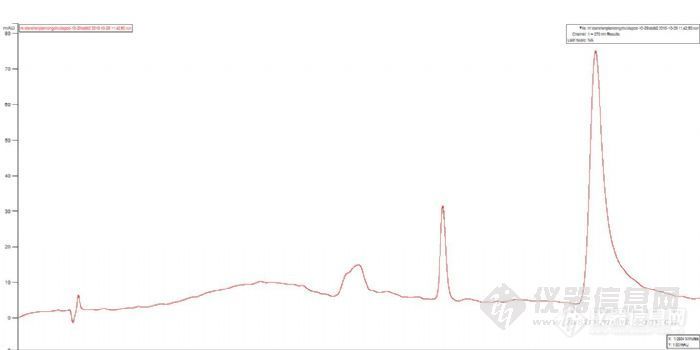
3、样品问题。把7个样品放入紫外下检测,看是否出峰。如果没有出峰,说明是样品处理出了问题。如果出峰,说明是液相出问题。
4、紫外没有出峰。检查样品处理过程,盐酸的量,浓度,PH和玻璃仪器的清洁度,以及溶出仪的有关参数。
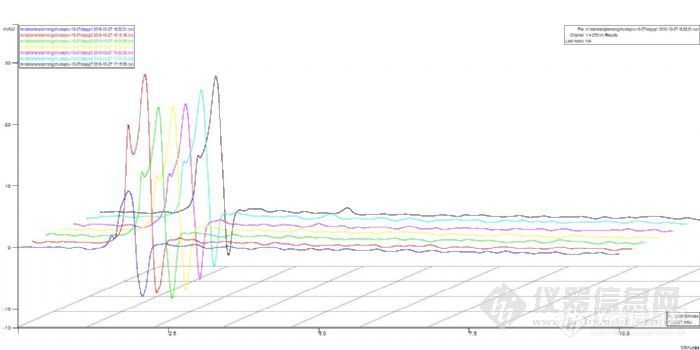
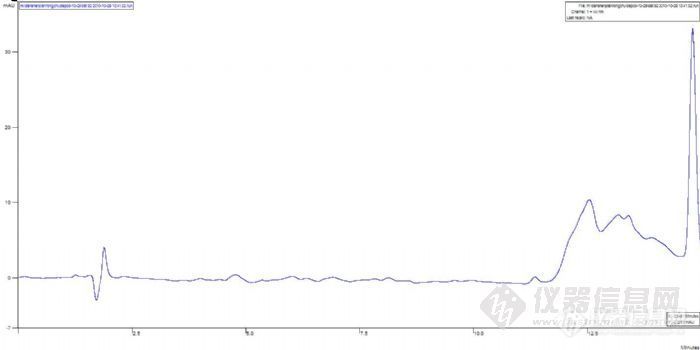
5、如果紫外出峰了,可能的原因是样品在酸性条件下解离,结构异构化,导致出峰时间延迟。延长分析时间或使用梯度洗脱,看是否有样品出峰。
阿三
第9楼2010/10/29
第5点中谈到解离,如果如此,出峰会提前,没有保留的,我觉得可能分析时间不够或如前面所说没有泵入流动相

































