气相色谱(GC)
八毛的老公
第1楼2011/12/12
我也在学习标准加入法的相关知识
xiaoxin2809
第2楼2011/12/12
关于第一个问题,我越来越觉得它是个数学问题而不是分析问题=.=
阿宝
第3楼2011/12/12
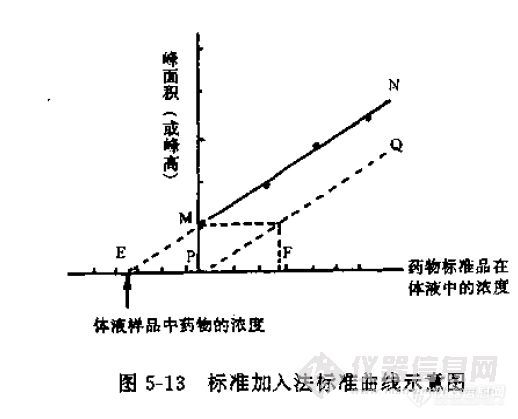
对于那个斜率问题,首先要明白斜率和什么因素有关?色谱条件、仪器状态稳定后,工作曲线的斜率就确定了
诗诗思思
第4楼2011/12/12
当杂质不影响出峰就可以用标准加入法。
安平
第5楼2011/12/12
斜率不变,好像没有依据。
p3329606
第6楼2017/11/14
楼主,你的问题解决了吗,最近也在准备做标准加入法的方法学,困扰中,还望楼主赐教
品牌合作伙伴
执行举报