液相色谱(LC)
老多_小多
第1楼2015/03/17
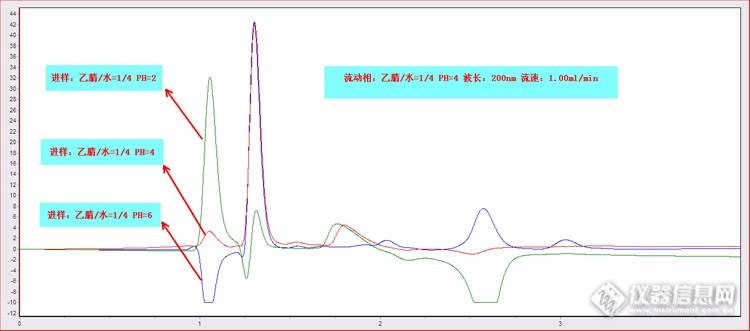
我认为200nm测定有很大影响,同样的色谱图,估计换个254,也许会完全不一样
orange85
第2楼2015/03/17
对pH,首先要注意色谱柱的pH使用范围,这是色谱柱使用基本保护;从谱图上看,pH为4时和流动相接近,所以色谱图好点,但也有好多峰啊,可能是色谱柱有关系,如果是脏了,得好好冲冲柱子。
夏天的雪
第3楼2015/03/17
1,低波长检测时流动相或者样品中的杂质可能被检出,显得杂质比较多,2,ph越低1.05min时的峰越高怀疑与加入的磷酸有关,3,后面倒峰的出现可能也和磷酸含量有关,之前有过误操作,测乙酸含量时用乙酸调了ph值结果出现负峰
vm88
第4楼2015/03/18
出负峰与PH有关系吗?
武灵
第5楼2015/03/18
1.05min处是否应该理解为死时间出的峰?
一片枫叶
第6楼2015/03/18
死时间出峰正负怎么理解呢?
第7楼2015/03/18
254nm的波长是能够得到解决的。
第8楼2015/03/18
色谱柱的因素是要考虑,进样组分与流动相组分的PH值差异,造成进样组分在固定相与流动相之间吸附与脱吸附的瞬间改变致使产生了正负峰应该是主要原因。
第9楼2015/03/18
固定不变的峰可能是进样组分的低波长杂质峰。正负变化的峰应该是其它原因引起的峰吧。
第10楼2015/03/18
不同的PH值有着不同的极性,不同极性的进样组分在色谱柱中,可能会造成吸附与解吸附短时间的变化,反映到检测器上可能就出现了正负峰。
品牌合作伙伴
执行举报