gc-MS可同时完成待测组分的分离、鉴定和定量,因此被广泛应用于复杂组分的分离与鉴定。并且也简化了样品的前处理过程,使得样品分析更简便。相比起色谱,实验员们在面对gc-MS时总是会觉得有点力不从心,今天小编就摘取一些来自前辈们总结的气质使用经验,让你在面对常见gcMS的问题时能够做到游刃有余!
别急,先说说液质联用与气质联用的区别
气质联用仪(gc-MS)是最早商品化的联用仪器,适宜分析小分子、易挥发、热稳定、能气化的化合物;用电子轰击方式(EI)得到的谱图,可与标准谱库对比。
液质联用(LC-MS)主要可解决如下几方面的问题:不挥发性化合物分析测定;极性化合物的分析测定;热不稳定化合物的分析测定;大分子量化合物(包括蛋白、多肽、多聚物等)的分析测定;没有商品化的谱库可对比查询,只能自己建库或自己解析谱图。
gc-MS能做什么?



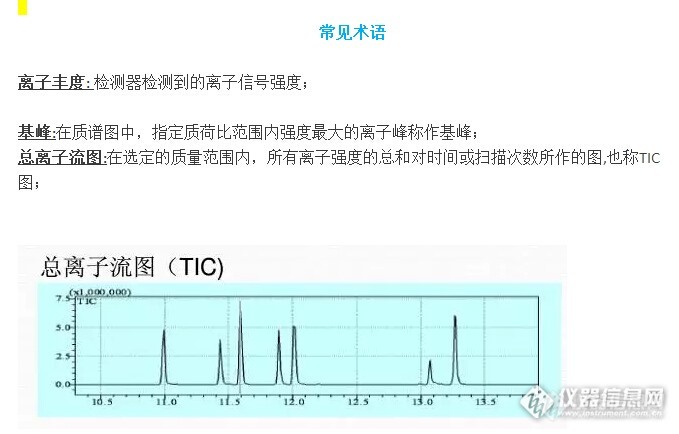
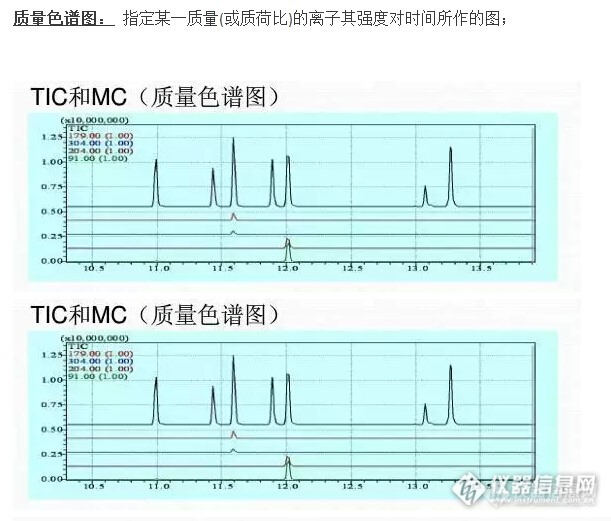
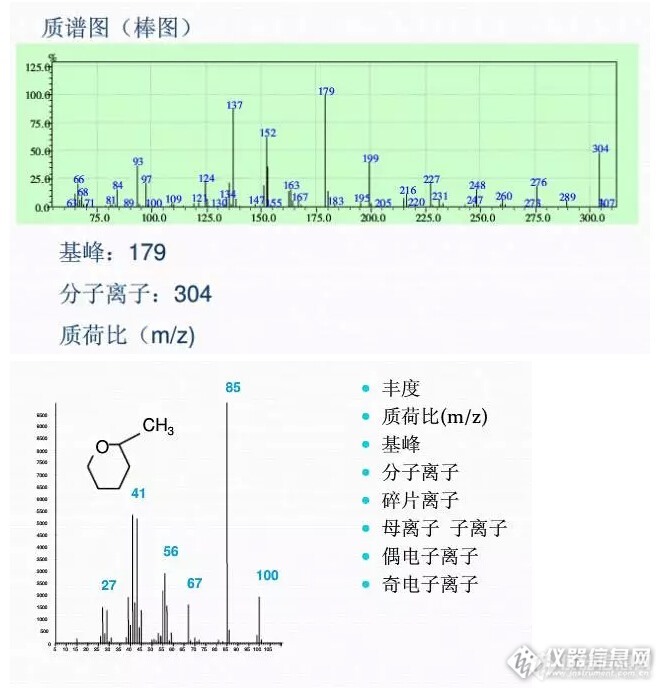
如何解质谱图:
(1)确定分子离子,即确定分子量 氮规则:含偶数个氮原子的分子,其质量数是偶数,含奇数个氮原子的分子,其质量数是奇数。与高质量碎片离子有合理的质量差,凡质量差在3~8和10~13,21~25之间均不可能,则说明是碎片或杂质。
(2)确定元素组成,即确定分子式或碎片化学式 高分辨质谱可以由分子量直接计算出化合物的元素组成从而推出分子式 低分辨质谱利用元素的同位素丰度。
(3)峰强度与结构的关系 丰度大反映离子结构稳定 在元素周期表中自上而下,从右至左,杂原子外层未成键电子越易被电离,容纳正电荷能力越强,含支链的地方易断,这同有机化学基本一致,总是在分子最薄弱的地方断裂。
不同类型有机物有不同的裂解方式 相同类型有机物有相同的裂解方式,只是质量数的差异需要经验记忆。
质谱解析的一般步骤
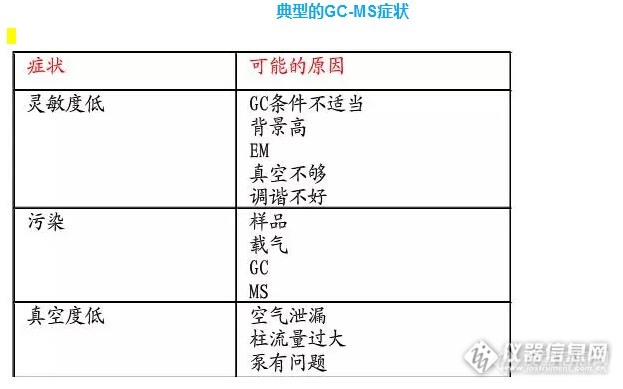
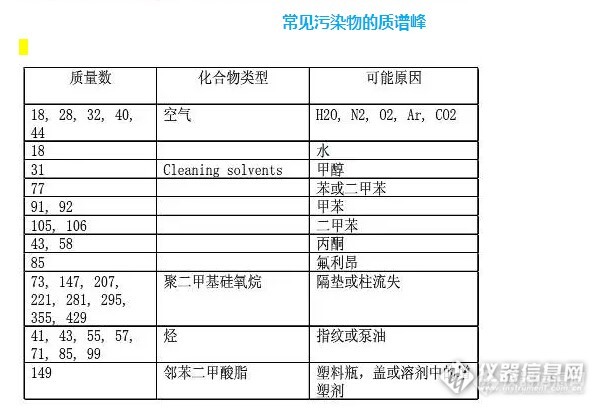
(1)核对获得的谱图,扣除本底等因素引起的失真,考虑操作条件是否适当
(2)综合样品其他知识:例如熔点,沸点,溶解性等理化性质,样品来源,光谱,波谱数据等.
(3) 尽可能判断出分子离子。
(4) 假设和排列可能的结构归属:高质量离子所显示的,在裂解中失去的中性碎片,如M-1,M-15,M-18,M-20,M-31......意味着失H,CH3,H2O,HF,OCH3......
(5)假设一个分子结构,与已知参考谱图对照,或取类似的化合物,并作出它的质谱进行对比。

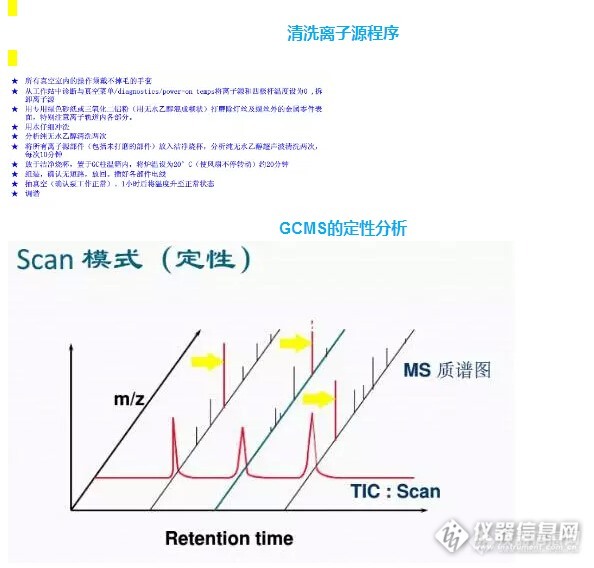

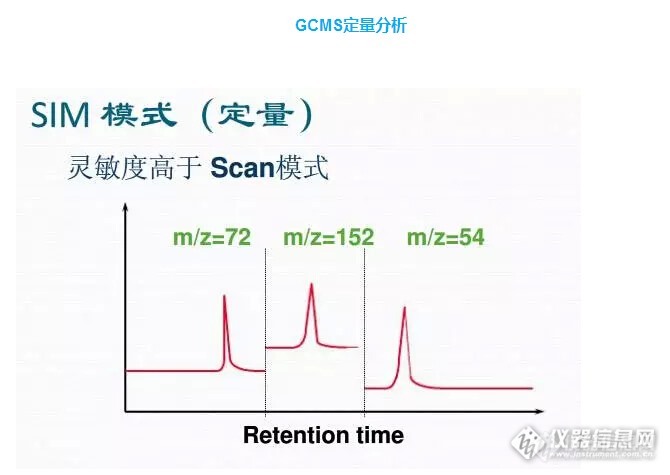
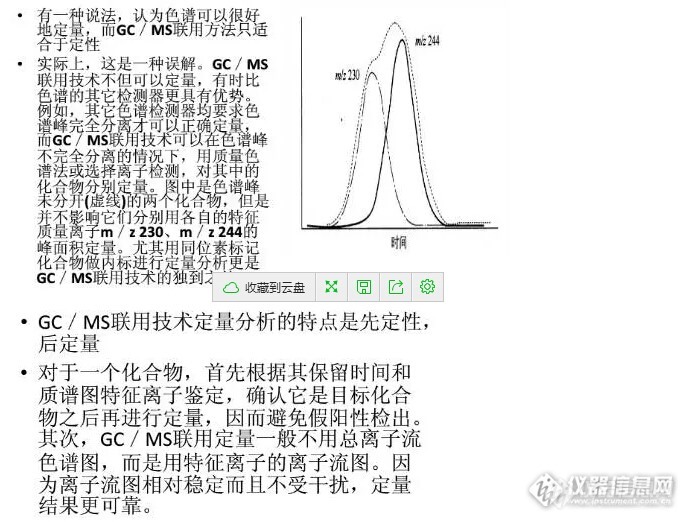
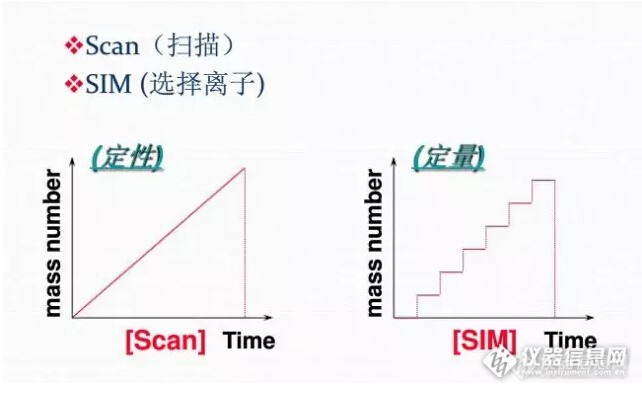
gcMS联用常用的定量方法和色谱一样,有归一法、外标法、内标法等。各有优缺点和使用范围。根据不同要求选择合适的定量方法非常重要。定量方法中响应值的计算可以用峰高法也可以用峰面积法,要看在线性范围内哪个测量的准确性和重复性更好。
定量分析中,如何选择参考离子呢?
gcMS定量中,一般都选定一个定量离子,然后再加上一个或多个参考离子来辅助定性。是否参考离子越多,定量越准?
参考离子的选择,一是要选一些丰度高,有一定辨识度的离子,二是要考虑前后两个峰的情况,选同一离子,方便分组。一个定量离子,三个参考离子。
参考离子越多,“定性”越准(几个离子,是没法准确定性的,只是说参考离子越多,越容易识别是否为目标物),但是每一个离子的扫描时间变短,对定量不利。
定性离子一般选2-3个,要是有标准提供定性,定量离子,一般根据标准确定,如果没有标准的选择丰度最大的离子做定量离子(无干扰),丰度2-4的做定性离子。
参考离子多,确实有助于判断峰。要得到更多信息,还是需要看SCAN得到的MS图啊。
如果用两个参考离子的话,可能会有一个参考离子未检出。一般SIM要得到比较好的峰型及准确结果,每个离子扫描驻留时间是有最低规定的,当选择离子过多,分配到每个离子的驻留时间就会相应降低,最终影响分辨率。
gcMS定量使用内标法还是外标法?
外标在定量过程中建立校正曲线是关键因素。是在特定的仪器和固定的实验条件下,加入一个给定的化合物,绘出浓度/含量和响应值(面积)和关系曲线。通过已知量样品,用积分法测定用是定量的峰面积,然后建立浓度/含量各峰面积的关系,再用这个曲线来外理未知样品。但要是实验仪器条件改变,这曲线就没有意义了。要重新建立校正曲线。
内标是采用加入已知道定量组分做归一化因子,其克服了外标法的缺点。但已知组分的内标物在校正样品和未知样品中最好加放相同量。这个如果是手动加放内标量,要想相同,有点难度。不过还是可以的,要多做就会好的。但内标物选择是有条件的。对于同一个项目可以永久使用,重要的是做样时,你要保证加的准。
样品中加入内标物,利用被测物与内标物校正因子的比值不变来进行定量的。 首先用被测物标准品和内标物配制标准溶液,求得定量响应因子或校正因子。
选择内标物的标准:
1. 样品中不存在; 2. 化学性质与样品相似;3. 与样品有相同的浓度范围; 4. 不会与样品发生反应; 5. 在感兴趣组分附近流出; 6. 可得到分离良好的、干净利落的峰;7. 色谱性质稳定; 8. 可迅速容易得到。



































