推荐厂家
暂无
暂无






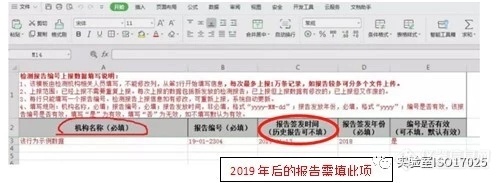
[align=left]内容来自网络。[color=#000000][b] 2019年 CMA年报解读[/b][/color]《市场监管总局办公厅关于开展2018年度认可及检验检测服务业统计工作的通知》:为加强检验检测机构监管,便于社会查询和监督,各检验检测机构应在检验检测统计直报系统上传本机构2016-2018年度出具的全部有效检验检测报告和证书编号,并于2019年起,每季度后一个月内报送上一季度出具的全部有效检验检测报告和证书编号。市场监管总局将据此向社会提供检验检测报告和证书编号有效性的网上查询平台。针对以上通知内容,相比较2018年年报增加了机构出具检验检测报告和证书信息的上报任务。各机构2019年提交年报将意味着需要上传本机构出具的全部有效检验检测报告和证书编号,如何填表,有哪些注意事项,以下示例为大家解答,供大家参考。1、下表中机构名称如何填写?答:填写检验检测机构自己的名称,不需要填委托单位名称。必须填写出具的报告编号,和报告签发年份。2019年后出具的报告和证书每一季度必须上报报告签发时间。[/align][align=left][img=,503,183]https://ng1.17img.cn/bbsfiles/images/2019/02/201902261024522763_1873_1619176_3.jpg!w503x183.jpg[/img]2、行业分类代码填写7450,系统显示为不合法的,如何解决?答:新版GB/T 4754—2017国民经济行业分类已将“检验检测”改成“7452”。[/align][align=left][img=,458,183]https://ng1.17img.cn/bbsfiles/images/2019/02/201902261026129203_6063_1619176_3.jpg!w458x183.jpg[/img][/align][align=left]3、如我方都为单位委托的检测,个人委托检验检测报告应该选什么?答:应该选择数值为0,但现在无法选择,需要等待系统更新后便操作。[/align][align=left][img=,418,57]https://ng1.17img.cn/bbsfiles/images/2019/02/201902261026506300_57_1619176_3.jpg!w418x57.jpg[/img]4、企业将检验检测业务分出来,成立了新的子公司,新公司是独立法人的,那原来注册的企业帐号今年起就不上报了,要注销,企业是否可以自己在系统中注销?答:不可以,只能由管理单位进行注销操作。[/align][align=left][/align][align=left]5、证书编号与发证机关不一致,不给保存,请教如何处理?答:按照163号令的规则填写证书编号,编号中间不要带空格。[/align][align=left][img=,510,271]https://ng1.17img.cn/bbsfiles/images/2019/02/201902261029301750_9704_1619176_3.jpg!w510x271.jpg[/img][/align][align=left]6、报告上传后如发现错误,如何进行修改操作?答:两个方式:1、从新excle导入,系统根据检测报告编号进行覆盖;2、在页面上进行修改。[/align][align=left][img=,474,217]https://ng1.17img.cn/bbsfiles/images/2019/02/201902261031031713_6407_1619176_3.jpg!w474x217.jpg[/img][/align][align=left]7、如检验检测机构同时获得国家级CMA,还有省级CMA。往年统计直报,都是以机构总体情况进行上报的国家级CMA,但是今年又新增了上报报告编号。在上报编号的过程中,发现机构选择这个框里,只有这一个选项,没有其他省级机构的名称,请问我们省级机构发的那些报告编号需要报送吗?答:可以上报,在上报excle中的“机构名称”中填写省级机构的名称。[/align][align=left][img=,402,95]https://ng1.17img.cn/bbsfiles/images/2019/02/201902261031516413_397_1619176_3.jpg!w402x95.jpg[/img][/align][align=left][/align][align=left][/align]
请教专业人士:我们公司的测试中心,也是通过CNAS认可的,想问下,如果公司内部签发的报告,审核及批准可以不用授权签字人签字么?如果可以不用的话,需要再对其他人任命么?
如果某一位授权签字人在批准人处签发的报告的量过少,会不会影响其持续能力(授权签字人能力)?

 400-892-9759
400-892-9759
 留言咨询
留言咨询
 留言咨询
留言咨询
 留言咨询
留言咨询










 我要推广仪器
我要推广仪器
 下载APP
下载APP