皮皮鱼
第1楼2009/07/01
取样我们仅仅从安全性上讨论一下,下面继续讨论进样的问题。关于取样的深入讨论,有相关经验的朋友可以在后面讨论,或者开一个新的帖子。
由于常温差压下是气体,因此液化气体样品既可以气化后气相进样,也可以直接液相进样。早期标准都是选用气化后进样的方法,一般都是钢瓶倒置,下面连接一段不锈钢盘管,泡在水浴槽内,后面连接定量环进样口。理论上来说,这相当于气相进样了。但这里要讨论一下气化过程。这一气化过程实际上存在很多可能的问题,最关键的问题就是轻组分气化快,重组分气化慢,最终导致进入定量环的气体,无法代表液化气体的真实情况。
要保证气化后气体对原来液体的代表性,必须保证以下几点:
1、样品以液态流出取样容器。钢瓶的话,应该用没有溢流管的一端向下,从下面放出液体去气化装置。
2、从容器中放出的液体,必须在气化装置中得到完全、快速、充分的气化。为了控制气化后气体的流速,容器出口的节流阀非常关键,要能够且很方便的调节流速。
3、整个气化过程中不能有污染物进入、不能有死体积、不能有对样品中关心组分的吸附等问题。
事实上,仅凭钢瓶出口针型阀很难做到以上第2点,经常是开大一点气流速度就过快,小一点就没有气体流出。特别是在气化温度较低,气化不够迅速的条件下,经常开一下钢瓶阀,气流慢慢的增加到不能接受,然后赶紧关死阀门,气流还要慢慢降低,过一段时间才会完全没有气泡发生。这种情况下,气体对液化气体的代表性是非常差的,这样的进样情况,几乎无法得到正确的分析结果,也无法保证分析的再现性。因为这个情况下,先流出来的是液化气体样品中的轻组分,后流出来的是样品中的重组分,进样阀切换时间的不同,就会带来结果的不同。
为了解决这个问题,一些厂家开发了专用的气化装置,一些厂家干脆放弃了液化后进样,而是采用了直接液体进样。从经验上看,我个人倾向于用液相阀进样,因为这个进样方式可以肯定具有足够的代表性。
皮皮鱼
第2楼2009/07/01
液相进样阀实际上就是两小段管线,在阀门内来回切换。当我们用液化气体样品充满一段管线,然后切换到载气线路中时,这一段管线中的样品以液体状态进入色谱系统,并在载气带动下气化并转移到色谱后系统。
液相阀进样可以确保不会发生气化过程中的不同组分气化速度不同造成的问题。因为即使发生气化速度的差异,样品也已经到了色谱系统中了。但液相阀有自己本身的问题。
液体气化体积增加是大的。液体气化后变成气体的体积是原来液体体积的倍数称为气化比。一般液体在1个大气压下的气化比在150-10000倍。因此为了确保进样量合理,液相阀需要有一个非常小的体积,一般只有0.2ul左右。这样小的体积,造成阀门中定量的这一小段管线,要比外面连接管线更细,内径更小。这就导致液相阀存在的第一个问题:容易堵塞。如果样品不洁净,含有小的固体颗粒,这些固体颗粒很容易通过管线进入液相阀,并在液相阀内卡死。因此要在液相阀前管线上加装过滤器。事实上我在应用过程中,即使有过滤器,也有堵死的情况发生。
液相阀的第二个问题是气泡。液相阀内只有0.2ul的体积,如果这一体积内存在气泡,则会导致进样量过小,产生很大的误差。为了确保阀内没有气泡,液相阀出口都装有调节流速的针型阀,通过控制流速,确保从针型阀喷出的是液体,来控制阀内样品保持液态。
液相阀的第三个问题是如果样品自身压力有限,如何保证样品在液相阀内的置换速度,能够做好充分置换并避免气泡?
液相阀的第四个问题是体积的稳定性。有时候需要外标法定量,这个时候如何保证液相阀内样品体积稳定?
液相阀的第五个问题就是这一体积如何确认并利用气体标准气来标定?我们知道液化气体标准样品是很难找到的。
事实上,解决这些问题的办法是为钢瓶提供背压。也就是说,从钢瓶的另一端,连接惰性气体钢瓶,接受来自钢瓶的压力。例如利用N2气钢瓶提供1.5MPa的压力给液化石油气样品来确保置换速度,避免气泡。
同时稳定的背压,可以确保液相阀内容积的稳定。液体本身具备不可压缩性,不同压力下的样品体积的差异并不大,但稳定的压力无疑可以确保进样体积的稳定性。
最后一个问题:如何利用气体标准样品来标定液相进样阀,外标法测定液化气体样品?做为一个留下来的问题,欢迎大家跟帖讨论一下。
felix126
第7楼2010/07/17
我在工作中终于用到液阀了,不过我们设计的背压气体是载气,就是用三通从载气管路上接过来的。我觉得万一采样人员心情好采的比较满或者没放空,钢瓶的压力大于载气气路的压力,这样样品会不会倒流污染载气呢?
还有如果液阀前端有阻尼而后端没有阻尼,这样的定量会不会有偏差呢?
关于用气体标样标定液阀,我觉得可样从GSV进样计算校正因子再利用液体膨胀系数来校正这个校正因子,瞎猜的啊 不知道对不对。
皮皮鱼
第8楼2010/08/22
工作比较忙,才看到你的回帖,回复晚了,原谅。关于你的问题:
首先,背压是载气这是不可以的。
原因:载气压力只有0.7MPa左右,很多液化气体压力高于载气压力,容易造成样品污染载气管线。就是不高也害怕啊。。。。。。何况这么低的背压,难以保证阀内是液体。还有,如果这边不小心把背压通大气了,会造成载气系统压力下降,也会影响所有相连的色谱仪。总之,效果没保证,危险非常大,因此不能这样做的。还是专门设置一条线路提供背压吧。
其次,液阀前端有阻尼,后端没有阻尼也是不可以的。
原因:这样这个阻尼没作用啊,或者说是起反作用的啊。阻尼的存在是为了保证阀内承受背压,因此样品保持液态。如果你阻尼在阀前,那么背压在阻尼处发生压降,阀内就是低压了,样品必然汽化啊。只有阀后装阻尼,背压在阀后发生压降,才能保证阀内的高压。
最后,好像关于外标定量,我在这个系列的后面帖子中说了,请查阅。
皮皮鱼
第9楼2016/02/01
好久不来,不知道讲过这个。但看了一下,当时忙乎,讲的很粗糙。这里详细说一下吧。
液化气体样品的气化进样
早期标准都是选用液化气体样品气化后进样的方法,一般可采用水浴加热气化和闪蒸仪气化两种方式。这两种气化方式的原理是相同的,其示意图如下: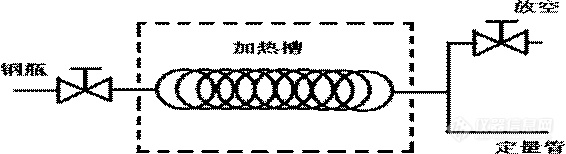
水浴加热气化进样具体操作为:将钢瓶倒置,下面连接一段不锈钢盘管,泡在水浴槽内,后面连接定量环进样口。
闪蒸气化进样具体操作为:将钢瓶倒置,下面出口端钢线与闪蒸仪进口连接,经闪蒸仪加热气化后,闪蒸仪出口连接定量环进样口。
理论上来说,液化气体的气化后进样,当样品从气化器出口出来,已经相当于气体样品进样了。但气化后进样与气体样品进样相比多出一个气化过程,这一过程的操作不当,将带来很多问题,影响最终结果。其中,这一过程最关键的问题就是在气化过程中,由于沸点关系,造成轻组分气化快重组分气化慢,最终导致进入定量环的气体,与液化气体样品的真实成分出现差异。
要保证气化后气体能够代表原来液体样品的实际情况,进样时必须保证以下几点:
样品以液态流出取样容器。钢瓶应该用没有溢流管的一端向下,从下面流出液体去气化装置。绝不能让钢瓶正立,从钢瓶上方取气体进样分析。
皮皮鱼
第10楼2016/02/01
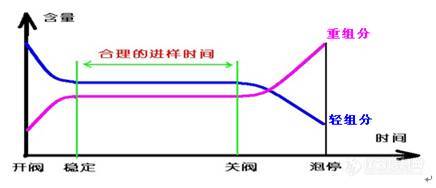
考虑到轻重组分在闪蒸仪中气化情况不同,分析各组分在闪蒸仪中气化过程应如下图所示。
|
|
| 图12-3 闪蒸气化进样原理图 |