chemistryren
第1楼2009/08/08
1.你说的增益是不是就是负高压?如果是,那这点也是正常的.氢化物发生我没做过,不过估计你的空白这点吸光度属于正常范围.并不高.
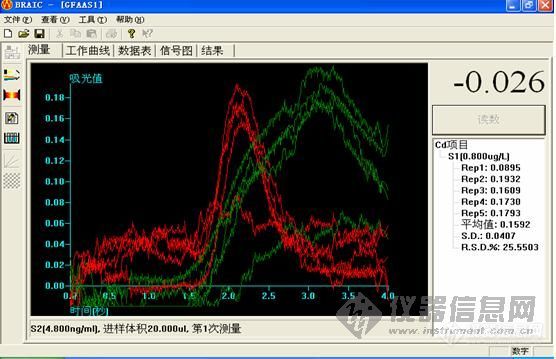
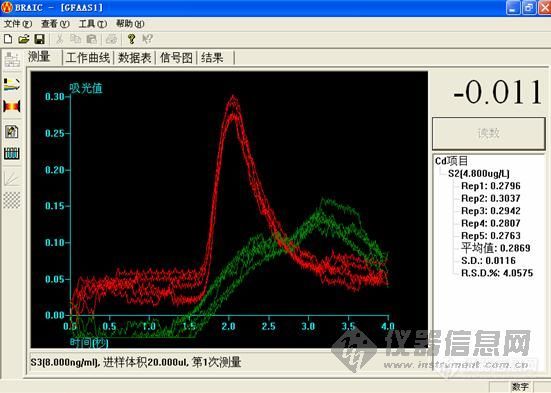
2.镉是用石墨炉测的还是用氢化物发生测的?用石墨炉测的话有没有用基改?
3.两者是混在一起的.
4.你在引号之前用了什么试剂?有硫酸或别的酸吗?
flytiger603
第2楼2009/08/08
1.增益是负高压,但是两个定义应该不太一样。关键是汞灯的值在不停的跳,是不是灯不稳啊?我怕这样测,会使吸收值的误差很大
2.镉使用石墨炉做的。基改是什么?
4. 引号中是药典上的消解方法,之前就是用硝酸-高氯酸(4:1)浸泡过夜就行了。没有用别的溶剂。
flytiger603
第7楼2009/08/10
2 As 的分析 (灯的增益值为420)
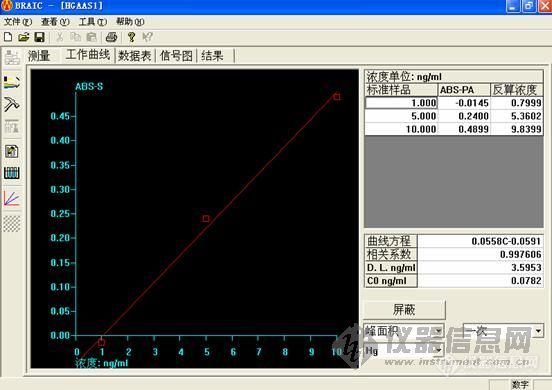
用翰时的氢化物,一切都是按照药典上的要求:分别精密量取砷标准储备液适量,用2%硝酸溶液制成每1ml分别含砷5ng、20ng、40ng的溶液。分别精密量取10ml,置25ml量瓶中,加25%碘化钾溶液(临用前配制)1ml,摇匀,加10%抗坏血酸溶液(临用前配制)1ml,摇匀,用盐酸溶液(20→100)稀释至刻度,摇匀,密塞,置80℃水浴中加热3分钟,取出,放冷。取适量,吸入氢化物发生装置,测定吸收值,以峰面积(或吸光度)为纵坐标,浓度为横坐标,绘制标准曲线。
检测:采用适宜的氢化物发生装置,以含1%硼氢化钠的0.3%氢氧化钠溶液(临用前配制)作为还原剂,盐酸溶液(1→100)为载液,氮气为载气,检测波长为193.7nm,背景校正为氘灯。
但是,标曲做了三个点,都没有吸收都如图: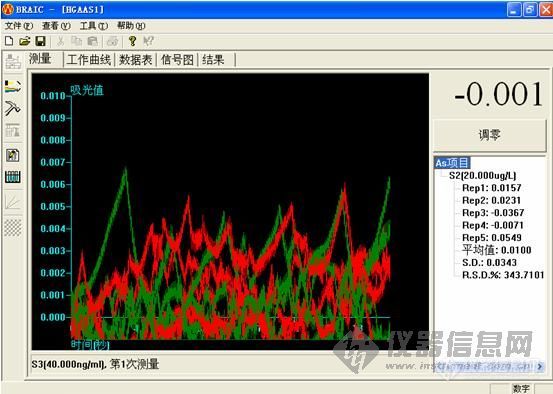
打电话给他们的,说是标准品用2%的硝酸做不出来,用他们给的方法,要用10%的盐酸。我还没有试。如果真的是按照他们说的,那药监局来检查,我们应该怎么交代,没有按照药典做,出来的结果,他们认可吗?
3.Hg 的分析 (灯的增益值为197,但是,校正用氘灯的能力上不去,只有20%)
检测:采用适宜的氢化物发生装置,以含1%硼氢化钠的0.3%氢氧化钠溶液(临用前配制)作为还原剂,盐酸溶液(1→100)为载液,氮气为载气,检测波长为193.7nm,背景校正为氘灯。
1.0ng/ml 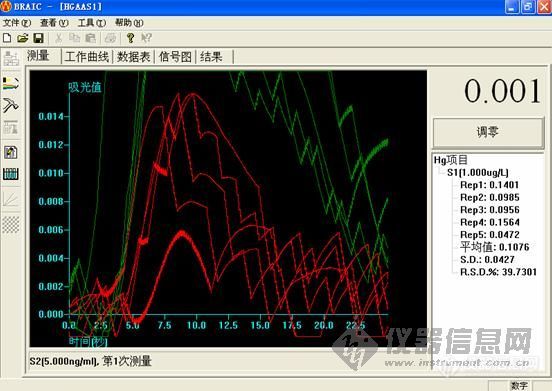
5.0ng/ml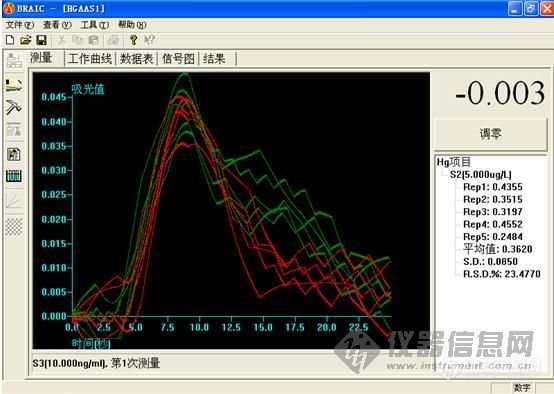
10.0ng/ml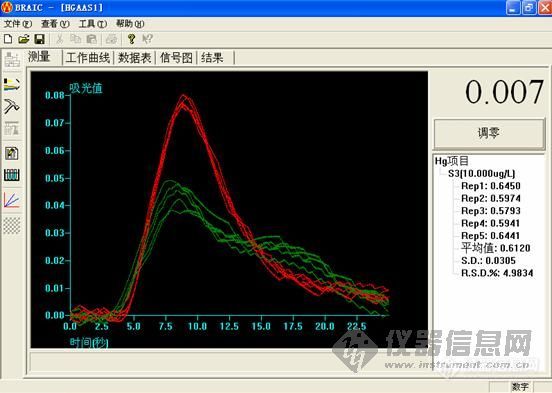
标曲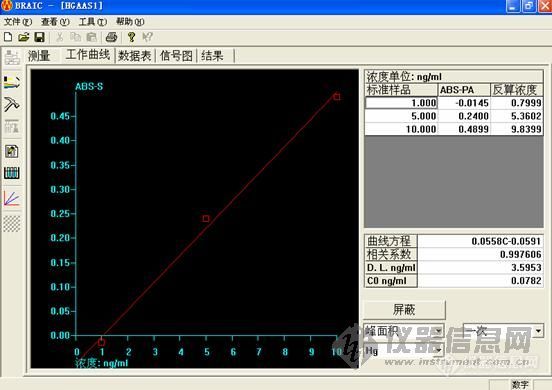
这个做下来,低浓度的时候很难看,高浓度的时候还可以。工程师说,用的还原剂有问题,浓度太小,根据他说的,都不按药典说的来。
大家先帮我看看,是我仪器的问题,还是灯,或者是方法的问题,我好根据改进。
谢谢大家!
purplehap
第8楼2009/08/24
[div][/div]
1.增益是负高压,但是两个定义应该不太一样。关键是汞灯的值在不停的跳,是不是灯不稳啊?我怕这样测,会使吸收值的误差很大
2.镉使用石墨炉做的。基改是什么?
4. 引号中是药典上的消解方法,之前就是用硝酸-高氯酸(4:1)浸泡过夜就行了。没有用别的溶剂。
你所说的汞灯值在不停的跳是什么意思?是不是能量平衡后(假如说平衡到50%),但是在50%左右不停的变化?如果是的话,你可以有2个办法看到底是哪里的问题:
1.多寻峰几次,看波长重复性,如果重复性不好,那么就是你的波长机构(不知道瑞利用什么波长机构)部分的问题,你要找仪器厂商
2.如果波长重复性好,那就是你的电路问题,还是找厂家