

庐山居士 2010/07/15
[color=#0021b0][size=3]楼主的[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原吸[/color][/url]应该是瓦里安的[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原吸[/color][/url],单石墨炉,塞曼扣背景,应该有自动进样器吧,本人也是瓦里安的用户,有几项建议,楼主可以尝试一下: 1、改变Pb测定波长,一般都用283.3nm,你改成217nm尝试一下。 [img]https://ng1.17img.cn/bbsfiles/images/2010/07/201007151600_230835_1630080_3.jpg[/img] 2、有版友提到提到灯插座的问题,我也遇到过,四个灯座使用时间长难免有接触不好的情况,我们就遇到灯没装好无法点亮的情况,还有灯使用时间长了,必须加大电流才能点亮,更换一下插座尝试一下。 [img]https://ng1.17img.cn/bbsfiles/images/2010/07/201007151605_230836_1630080_3.jpg[/img] 3、楼主在实验室对故障进行了多次尝试,石墨锥、石墨管、空心阴极灯都没多大问题,那就再检查PSD自动进样器吧,如果还有配件,就把进样器上的毛细管给换了吧,原来我们做血铅时,先开始很正常,过了一段时间后,吸光度突然下降,后来查了好久的原因,猜毛细管内壁有污染,剪掉前面一段后就恢复正常了。 [img]https://ng1.17img.cn/bbsfiles/images/2010/07/201007151612_230839_1630080_3.jpg[/img] 4、PSD自动进样器的注射器不要忽视,尝试多次清洗和排气泡,这个注射器不会被遗忘了吧! [img]https://ng1.17img.cn/bbsfiles/images/2010/07/201007151621_230840_1630080_3.jpg[/img] 5、自动进样器清洗液定期更换一下。 [/size][/color][img]https://ng1.17img.cn/bbsfiles/images/2010/07/201007151623_230841_1630080_3.jpg[/img] [color=#0021b0][size=3]6、楼主仪器的条件:283.3nm,5mA,增益60%左右。狭缝0.5nm(仪器默认),塞曼扣背景,0.8赫兹。原子化条件:85度——10s,95度——40s,120度——10s,400度——8s,2100度——2s,2300度——2s,灰化温度改到过500度,没有区别。从中可以看出,楼主所用空心阴极灯非瓦里安灯,因为瓦里安铅灯灯默认电流为10mA,瓦里安推荐灰化温度为600度,我们加基改剂的目的就是确定最佳灰化温度和原子化温度,其实瓦里安[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原吸[/color][/url]有表面响应学方法优化石墨炉条件这个独特功能,这项功能非常强大实用,不用为确定最佳灰化温度和原子化温度犯愁,你可尝试一下,下面第2张图是我们利SRM功能优化Pb元素石墨炉条件的结果。 [/size][/color] [img]https://ng1.17img.cn/bbsfiles/images/2010/07/201007151646_230844_1630080_3.jpg[/img] [img]https://ng1.17img.cn/bbsfiles/images/2010/07/201007151703_230846_1630080_3.jpg[/img] [color=#0021b0][size=3] (如果还不行,就要查塞曼扣背景了,可我们是氘灯扣背景,没用过塞曼扣背景,所以这里也不敢多说。)[/size][/color]
theo0504 2010/07/13
有可能是盛放试样的容量瓶没有洗干净 被污染了 我在做石墨炉pb时候经常遇到 后来我直接把瓶子泡进1:1硝酸里面 把石墨炉用的自动进样器的小塑料瓶也洗干净
nhp 2010/07/16
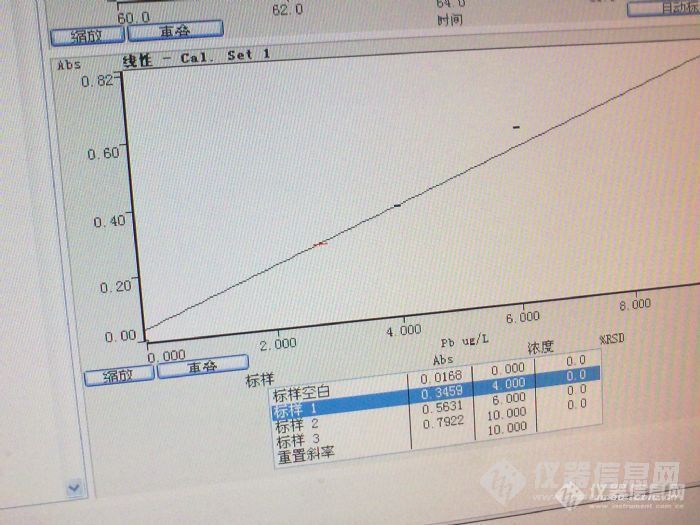
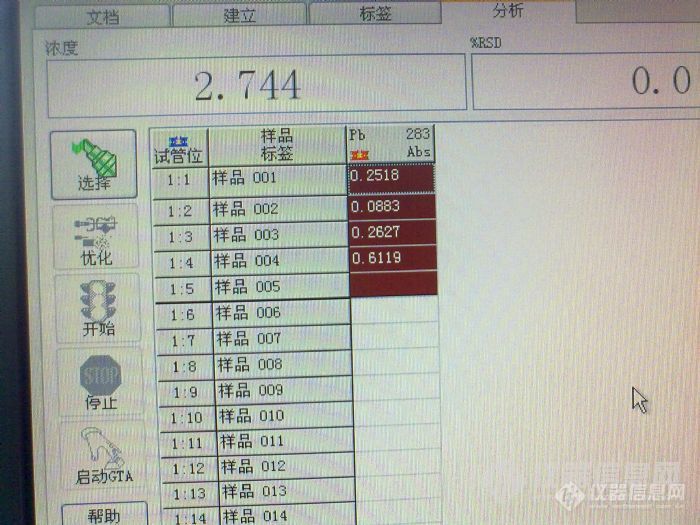
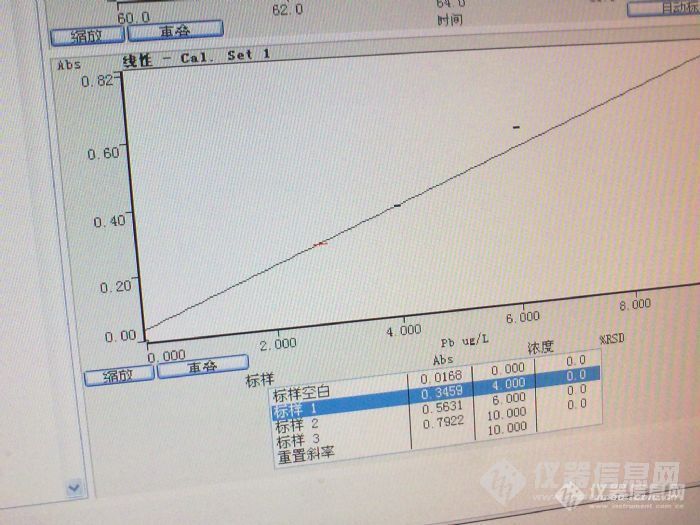
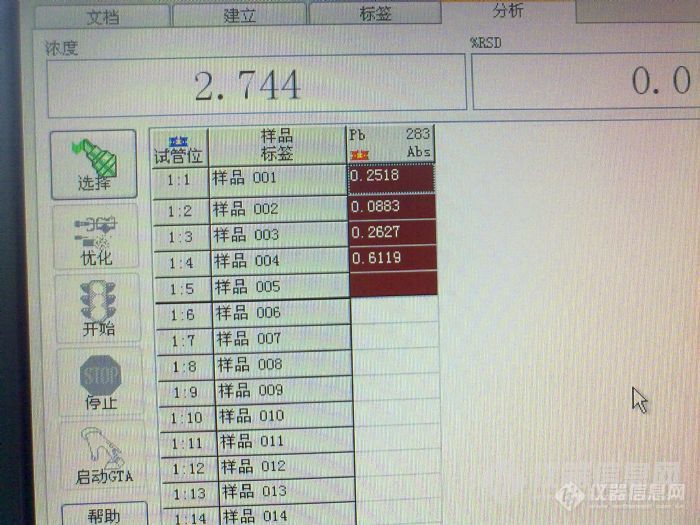
[quote]原文由 [b]邪龙妹妹(suminer)[/b] 发表: [size=4] 我这个帖子比较长,希望您能仔细阅读,找到问题,立马送分。 我们实验室用石墨炉测铅差不多五年了,斜率一直在0.007左右,即10ug/L的铅产生0.07的吸光度。空白从纯水的0.003到5%硝酸0.020,一直稳定。 最近仪器突然发疯了,10ug/L的铅吸光度可以做到0.79,斜率突然增高了十倍,我想难道是我的仪器灵敏度突然提高十倍?事实证明当然不是那么简单。当出现这种结果的时候,我们第一个反应是浓度肯定配错了,可能10ug/L的浓度配成了100ug/L,于是,和另一个同事一起,打开了一支新的标准,重新配置,在她的监督下,标准配置完全正确。再上仪器,吸光度仍然是0.79左右。 这时候,突然标准空白也变高了,吸光度能到0.26,空烧一下,再测空白,突然增大到1.3。在仪器网上看见有人说空白增大可能是石墨锥污染了,于是把炉体拆开,用无水乙醇擦拭N次,再用(20ml氨水+20ml丙酮+100mL水)擦拭,换一根新石墨管,空白仍然很高,而且不稳定,有时候到0.2,有时候又1.3。 强烈怀疑是空白污染,于是我们首先看是不是基体改进剂磷酸氢二铵污染,仅进基体改进剂,吸光度在0.02,跟以前相比,这个还算是正常;接着我们排除酸的污染,我们配置的空白是1%的硝酸,这个硝酸是分析纯的,几天前还用过的,一切正常。但是我们仍然决定换个酸试试看,于是换了一根石墨管,改用优级纯硝酸,问题一样,再重新拿了一年前不同批次的酸,问题仍然存在,最后还在商店里重新买了一瓶硝酸,上仪器结果还是这样。 没办法,只好干脆只进水了,标准溶液用纯水配置,石墨管也换了一根新的。进标准空白的时候还让我们喜了一下,因为空白降下来了,吸光度0.0084,再进标准的时候4ug/L的标准吸光度还是0.4左右,又做了6和10两个浓度,吸光度倒是成线性,0.995。心想只要成线性,就算是灵敏度越来越好了吧,于是开始进样品,放置了1%的硝酸和水作为样品,一看结果心又凉了半截,纯水吸光度不稳定,从0.08到0.8都有。 这时候我和同事又做了Cd和Ag两种元素,标准曲线和空白和以往的吸光度差不多,斜率也差不多,线性也很好,证明做别的元素都是对的。另外在做的过程中我们换了3支铅灯,每只灯的能量都是正常的。 最后一次进样,先进纯水(该纯水电阻值都在18以上),吸光度曲线不是尖锐的峰,而是M型双峰,空烧一次,再进样,吸光度1.4。工程师也问过了,只说是水和酸的问题,但水和酸我都换用过了,都没办法解决。最后没辙了,只得来求教各位达人了。 下面是我们做铅时候的吸光度: [img=653,365]http://ng1.17img.cn/bbsfiles/images/2017/10/201007091854498291_01_1786883_3.jpg[/img] 下面一个是1%硝酸及纯水的吸光度: [img=629,362]http://ng1.17img.cn/bbsfiles/images/2017/10/201007091854341851_01_1786883_3.jpg[/img][/size][/quote] 根据楼主的描述可能是污染问题,可以从试剂和容器上找原因,容器包括仪器进样杯和仪器进样部分。
wangxinxi7809 2010/07/12
这种问题查起来很麻烦,根据你的描述,我感觉是你的玻璃器皿出现污染,这样的话在检查仪器和标液都是没有用的,建议你把更换20%的硝酸,把玻璃器皿重新冲洗,浸泡24小时,容量瓶也要冲洗浸泡,应该能解决
zzs_71 2010/07/13
我看了你的仪器是varian的仪器,和我的仪器一样,按照你说的,你的石墨管是进口的还是国产的?我看了你的升温程序,我估计是你的升温的问题,你不加基体改进剂吧,主要是你还没有优化你的升温条件,另外你说你的原子化图形是双峰,更可以说明你的条件不对,说明原子化温度和时间不对,还有你的灯是进口原装还是仪器公司推荐的,灯电流有影响,你调一调试试,如果还有问题,请电话联系或者发邮件:zzs_71@126.com
hongwei-tlm
第1楼2010/07/10
从你描述的过程看,是先做完曲线再进1%HNO3和水,我认为你是不是应做完曲线后多清洗几次再进样分析?或空烧一下吹走残留?还有你是否观察进样毛细管头上是否有滞液?这种情况的很让人头痛,希望你尽快解决问题和大家分享
hongwei-tlm
第2楼2010/07/10
从你描述的过程看,是先做完曲线再进1%HNO3和水,我认为你是不是应做完曲线后多清洗几次再进样分析?或空烧一下吹走残留?还有你是否观察进样毛细管头上是否有滞液?这种情况的很让人头痛,希望你尽快解决问题和大家分享

































