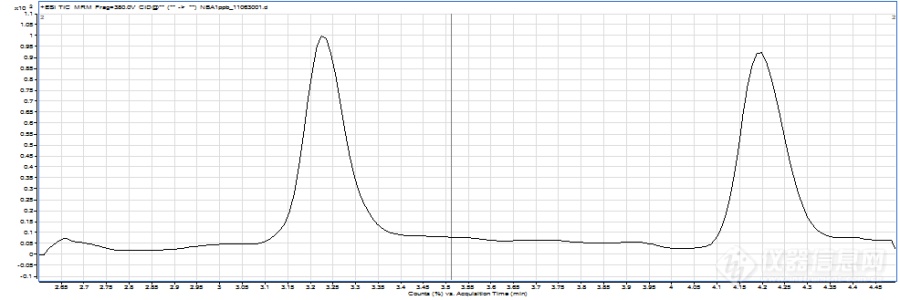
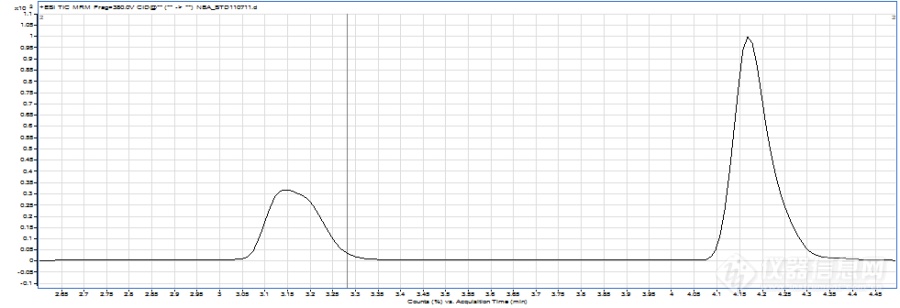
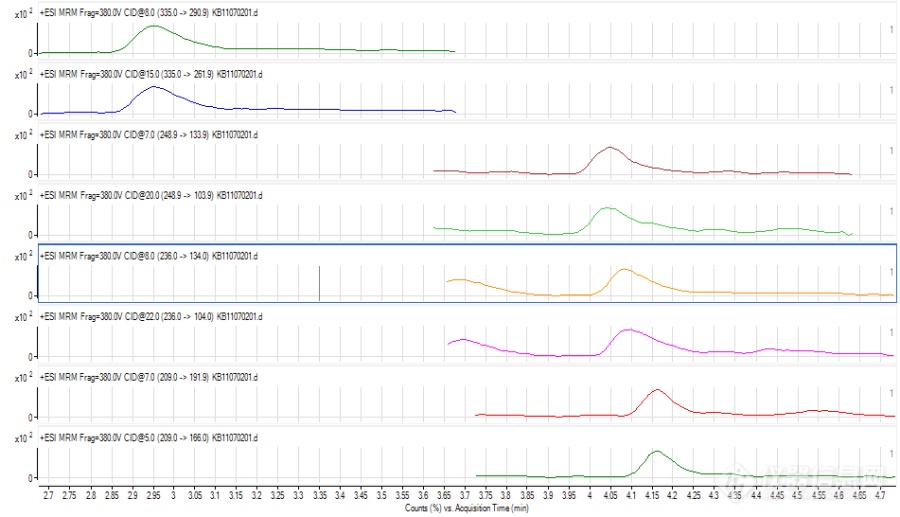
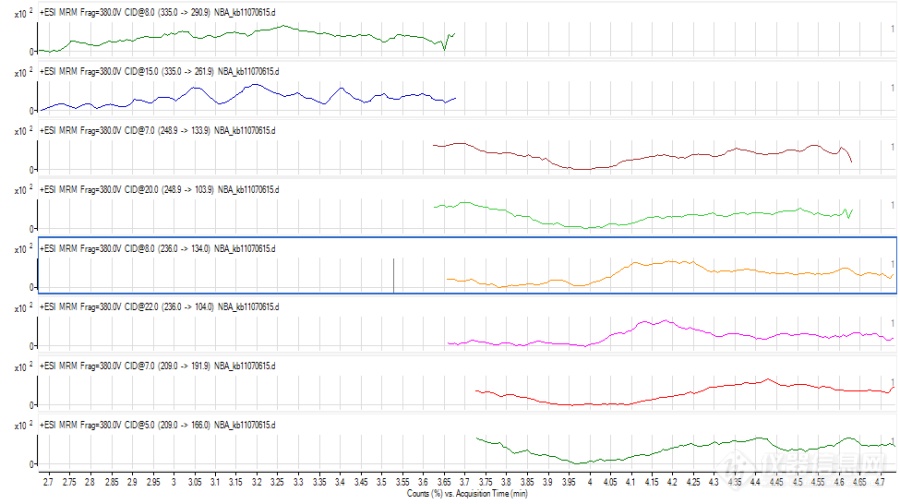
液质联用(LCMS)
yuduoling
第1楼2011/07/08
不错的经验,投你一票了
liumee
第2楼2011/07/10
青霉素类标准品好像不太稳定,我们的青霉素G经常就走不出来了,楼主有没有碰到类似情况?
coffee8
第3楼2011/07/10
为什么不同的仪器会有不同的峰至于容易出现加钠?加氢?加氨,还是加钾峰?这些与什么有关系呢?
happy爱米粒
第4楼2011/07/10
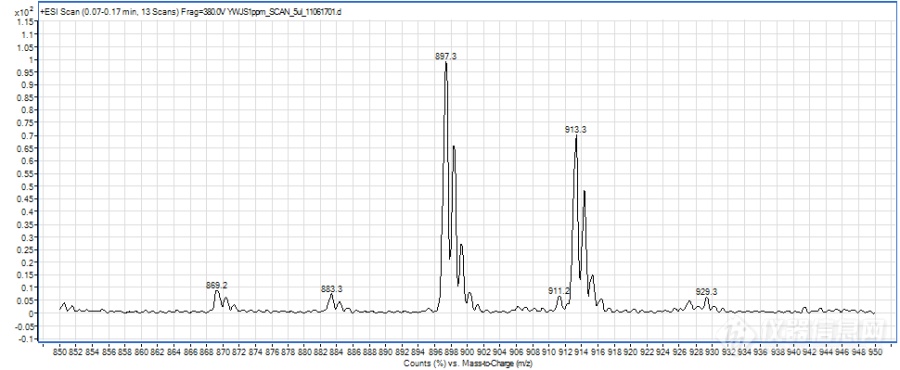
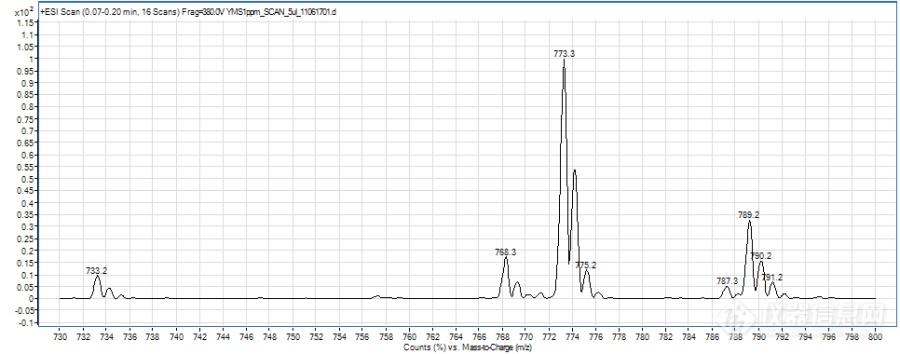
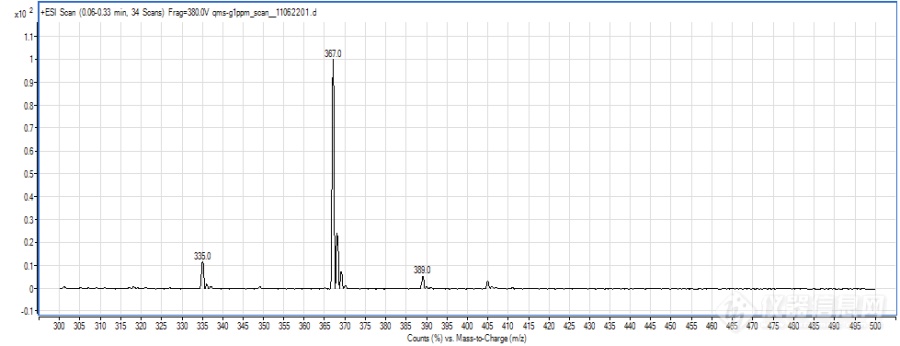
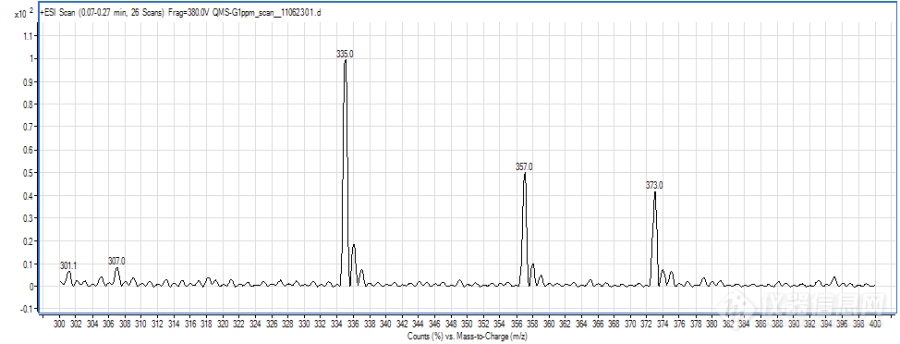
形成不同的分子离子峰可能除了跟化合物本身的性质有关,还与液相系统有一定的关系,因为是新买的液相,管路和试剂瓶中都会有一定钠、钾离子的存在,而premier液相系统相对使用时间较长,再加上流动相中使用了乙酸铵,抑制了加钠峰的形成。另,加钠、加钾峰太过稳定,不易碎裂,若碰撞能量过高,又会完全碎裂成小离子峰,所以能选择加氢峰、加铵峰的时候就不选择加钠峰,当然前提是要满足灵敏度的需要。
第5楼2011/07/10
青霉素类标准品是不太稳定,尤其是在酸碱环境中,我们配在乙腈+水=1+1里,还可以,有时候不一定是标准品分解了,也可能形成了其他的加合峰,可以做个MS2 scan看下。
第6楼2011/07/10
奇怪,怎么不能对本帖回复了?
dahua1981
第7楼2011/07/11
实际的方法转换很有学习意义
catty125699
第8楼2011/07/13
很有实际意义,学习了
流浪撒哈拉
第9楼2011/07/13
要学习学习
gys1982
第10楼2011/07/14
文章针对性太强了,很少人同时有以上两种设备,不过实验思路、想法值得学习,一看楼主技术很强,赞一个
品牌合作伙伴
执行举报