在线凝胶渗透色谱-气相色谱-质谱法测定茶叶中多种有机锡化合物残留
有机锡化合物(OTCs)是一类包含C-Sn共价健的化合物,主要用于聚氯乙烯聚合物的稳定剂、化学反应中的催化剂及农业生产中的除草剂、灭菌剂和杀虫剂[1]。由于有机锡化合物在工农业生产中的广泛应用,造成其在环境中的普遍污染,并沿着食物链的不断传递而对人体健康造成影响。作为一种重要的内分泌干扰物质[2],浓度在ng/L水平的有机锡化合物即可对水环境中的生物产生毒性危害[2,3]。因此,有机锡化合物的痕量与超痕量分析是当今环境和食品安全分析领域的重要研究课题。
本文采用在线凝胶色谱串联气相色谱-质谱(GPC-GC-MS)技术,结合大体积进样,同时检测茶叶中的6种有机锡化合物,具有检出限低、快速、简便、重复性好等优点及较好的实际应用价值。
1. 实验部分
1.1 仪器与试剂
GPC-GC/MS QP 2010 Plus在线凝胶渗透色谱-气相色谱质谱联用仪(日本岛津公司),配有程序升温(PTV)进样口和EI电离源;Envi-Carb活性碳固相萃取小柱(250 mg,3 mL,Supelco公司);Florisil固相萃取小柱(250 mg,3 mL,Supelco公司);快速混匀器(SK-1型,常州澳华仪器有限公司);氮吹仪(KL 5121型);低速离心机(10000 rpm以下,TDZ 4-WS型);电子分析天平(PB203-N型)。
丙酮、环己烷和正己烷均为HPLC级试剂;无水乙醚、H2SO4为分析纯;无水Na2SO4为分析纯,650℃灼烧4 h,贮于密封干燥器中备用。所用水均为超纯水。
六种标准品:二丁基二氯化锡(DBT)、三丁基氯化锡(TBT)、二苯基二氯化锡(DPhT)、三苯基氯化锡(TPhT)、三唑锡(Azocyclotin)和苯丁锡(FBTO)购自Dr. S. Ehernstorfer公司,纯度均大于95%。正戊基溴化镁(2.0 mol/L乙醚溶液),购自Aldrich公司。
1.2 色谱质谱条件
GPC条件:Shodex CLNpak EV-200凝胶色谱柱,150 mm×2.1 mm(i.d.);流动相为丙酮-环己烷(3:7,V/V);流速:0.1 mL/min;柱温:40℃;进样量:10 μL。GPC的淋出液收集时间为4.14~5.74 min。
GC-MS条件:惰性熔融石英毛细管柱色谱,5 m×0.53 mm(i.d.);DB-5MS预柱,5 m×0.25 mm(i.d.)×0.25 μm(膜厚);DB-5MS分析柱(25 m×0.25 mm(i.d.)×0.25 μm(膜厚)。
PTV进样模式:进样口初始温度120℃,保持5 min,以100℃/min程序升温至250℃,保持23.7 min。
色谱柱升温程序:色谱柱起始温度82℃,保持5 min,以20℃/min程序升温至250℃,保持5 min,再以30℃/min程序升温至310℃,保持10 min;载气:He气,纯度大于等于99.999%,流速为1.0 mL/min;电子轰击源(EI),70 eV;离子源温度:250℃;接口温度:280℃;扫描方式:选择离子监测模式(SIM)。
1.3 标准溶液的配制
分别称取100 mg(精确至0.1 mg) 各有机锡标准物于100 mL 容量瓶中,用甲醇溶解并定容,得1 .000 mg/mL有机锡标准溶液,于黑暗处4℃储藏,使用前用甲醇稀释成适当浓度的混合标准溶液。
1.4 样品前处理
称取0.5 g粉碎的茶叶样品于10 mL玻璃试管中,加入1 mL盐酸溶液(1 mol/L),混匀后加入1 mL丙酮,2 mL正己烷,于快速混匀器上振荡3 min,离心,用尖嘴吸管将上层清液移入另一试管中,下层残渣再用正己烷(每次2 mL)提取2次,合并提取液,用氮气吹至近干,加入少量无水硫酸钠,用2 mL无水乙醚分3次将残余物转移入具塞罗口试管中,加入0.8 mL 的正戊基溴化镁乙醚溶液(2.0 mol/L), 快速振荡1 min,放置30 min,于冷水浴中徐徐加入5 mL0.5 mol/L的硫酸溶液除去过量的正戊基溴化镁,再加入5 mL水,用6 mL正己烷分3次提取,合并提取液,用氮气吹至约1 mL。
将Envi-Carb活性碳固相萃取柱和Florisil固相萃取柱用树脂连接头上下连接起来,上面为活性碳小柱(其上覆盖约1 cm厚无水硫酸钠),先用6 mL乙醚正己烷混合液(5+95,V/V)预淋洗,活化小柱,然后将上述衍生化浓缩液上样,用6 mL乙醚正己烷混合液(5+95,V/V)洗脱。洗脱液用氮气吹至近干,用GPC流动相定容至0.5 mL,供GPC-GC/MS分析。
标准溶液用氮气吹干溶剂后,加入2 mL无水乙醚,按上述同样方法衍生、提取和净化。
2. 结果与讨论
2.1 前处理方法的选择
有机锡化合物常用的提取试剂有甲醇、丙酮等极性溶剂[4,5]和正己烷等非极性溶剂[6,7]。由于有机锡化合物一般具有较强的极性,当使用非极性溶剂提取时,常加入环庚三烯酚酮(tropolone)[8,9]、二乙基二硫代甲酸钠(DDTCNa)[10~12]或适当浓度的酸(如HCl、HBr等)[8,11,13,14],以提高提取效率。本实验在样品中加入盐酸,用丙酮-正己烷混合溶剂进行提取,方法简单,快速,提取效率高。实验比较了不同的盐酸浓度(0.0 mol/L,0.1 mol/L,1.0 mol/L和5.0 mol/L)对各有机锡化合物提取效率的影响,结果发现,加入低浓度的盐酸溶液(0.0 mol/L和0.1 mol/L)时,各有机锡化合物的提取效率较低,加入1.0 mol/L和5.0 mol/L的盐酸溶液时的提取效果均较好,因此我们选择1.0 mol/L的盐酸溶液。
由于茶叶中含有较多的色素,因此在样品净化过程中需使用一根活性碳小柱以除去色素。对于其它的干扰物质,我们比较了Alumina-N固相萃取柱、Florisil固相萃取柱和LC-Si固相萃取柱的净化效果,结果发现Florisil固相萃取柱的净化效果最好。所以,我们最终采用Envi-Carb活性碳固相萃取柱串联Florisil固相萃取柱作为净化手段,再结合在线凝胶色谱的进一步净化,去除了大部分基质干扰,使得我们在检测结果时能有很好的灵敏度。
2.2 GPC-气相色谱-质谱分析
在线GPC-GC/MS是将GPC净化系统与GC/MS分析检测在线连接起来,实现了从样品净化到分析检测的自动化。样品提取液经过GPC色谱柱的分离,通过阀切换,排出从GPC色谱柱洗脱快的油脂、色素等干扰组分,将目标分析物切入GC,经GC色谱柱分离后,在MS部进行检测。切入GC/MS的起止时间对于GPC的净化效果以及分析检测的灵敏度有重要关系。如果切入的起始时间过早或切入的结束时间过迟,则进入GC/MS系统的干扰物质增多,不仅不利于检测,而且对GC色谱柱以及MS系统不利。反之,如果切入的起始时间过迟或切入的结束时间过早,虽然进入GC/MS的干扰物质减少,但可能导致切入GC/MS的目标分析物的量减少,从而损失检测的灵敏度。我们通过实验,确定将GPC 4.14~5.74 min时间段的淋出液切入GC/MS系统,既确保有机锡化合物全部进入GCMS分析系统,又最大限度地利用了GPC净化的作用。
2.3 标准曲线与检出限
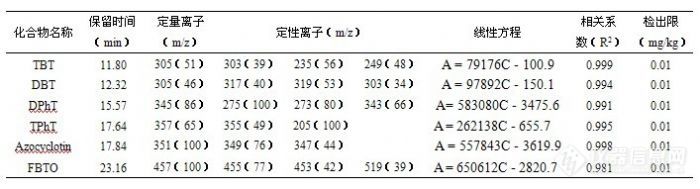
取一定量的6种有机锡化合物的标准混合溶液,用氮气吹干溶剂后,按2.4所述方法衍生、提取和净化后,用GPC流动相定容,并配制成质量浓度为0.01~1.0 mg/L的衍生化标准工作溶液,进行GPC-GC/MS分析,标准溶液的GC/MS色谱图见图1。以各衍生物的定量离子(定量离子和定性离子及其相对丰度见表1)的峰面积(A)对它们的质量浓度(C,mg/L)绘制标准曲线,并求得直线回归方程和相关系数,结果见表l。
在定量计算过程中,由于样品基质中的碎片离子可能与被测化合物的基蜂存在干扰,因此,为了保证定量的准确性,我们选择没有干扰或干扰最小的碎片离子作为定量离子。在定性确证时,被测物选择离子的相对丰度应当与浓度相当的标准工作溶液一致。定性离子相对丰度允许的偏差为:相对丰度>50%时,允许的相对偏差为±10%;相对丰度范围在20%~50%、10%~20%和<10%时,允许的相对标准偏差分别为±15%、±20%和±50%。
2.4 方法的检出限、回收率和精密度
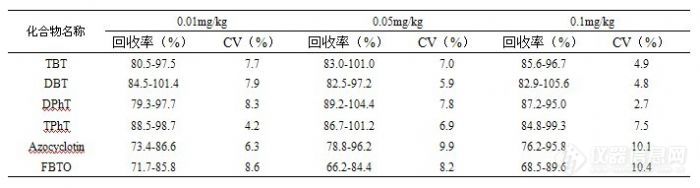
方法的检出限根据10倍信噪比来确定(见表1)。本方法采用标准加入法,取粉碎均匀的茶叶样品,准确加入6种有机锡化合物的混合标准溶液,分别制备添加浓度为0.01、0.05 和0.1 mg/kg的样品,按2.4所述方法进行样品处理,并进行GPC-GC/MS分析,每个添加水平重复测定6次(空白样品及添加样品GC/MS色谱图见图2),计算平均回收率和相对标准偏差,结果见表2。由表2可见,方法的回收率为66.2~105.6%,相对标准偏差在2.7~10.4%之间,满足检测的要求。
3. 结论
本实验采用在线凝胶色谱串联气相色谱-质谱(GPC-GC-MS)技术,结合大体积进样,建立了一个灵敏、快速、简便、重现性好的有机锡化合物的检测方法。该方法的精密度、准确度均符合要求,可用于茶叶中有机锡化合物的快速检测。

图1 6种有机锡化合物的选择离子流图(1.TBT 2.DBT 3.DPhT 4.Azocyclotin 5.TPhT 6.FBTO)
Fig.1 SIM chromatogram of 6 organotic compounds
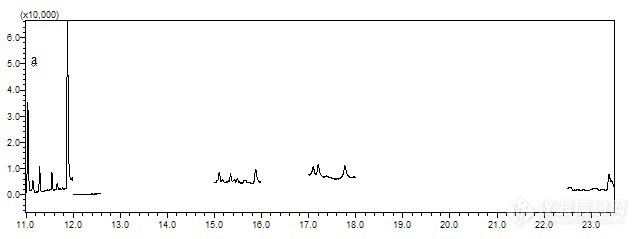
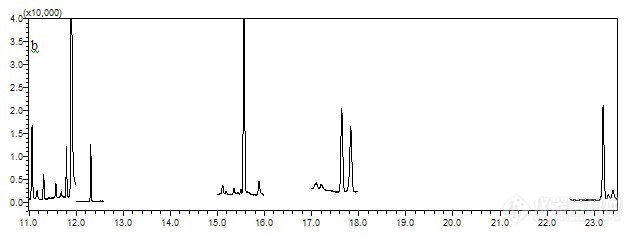
图2 空白茶叶样品(a)和添加样品(b)的选择离子流图
Fig.2 SIM chromatograms of blank tea sample and spiked tea sample
表1 6种有机锡化合物的色谱保留时间,定量、定性选择离子及相对丰度,线性关系及检出限
Table 1 Retention times,quantitative ions,qualitative ions and relative abundances,linear relationships,detection limits of 6 organotic compounds
表格 2 三种加标水平下6种有机锡化合物的回收率和相对标准偏差(n=6)
Table 2 Recoveries and RSD% of 6 organotic compounds at three spiked levels
参考文献
[1] 吴永宁,江桂彬. 重要有机污染物痕量与超痕量检测技术. 北京:化学工业出版社, 2006:459.
[2] K. Fent. Ecotoxicology of organotin compounds. Crit. Rev. Toxicol, 1996, 26: 1-177.
[3] R. J. Maguire. Environmental aspects of tributyltin. Appl. Organomet. Chem., 1987, 1(6): 475-498.
[4] I. S. Krull, K. W. Panaro, J. Noonan, et al. The determination of organotins (TBT) in fish and shellfish via gas chromatography-flame photometric detection and direct current plasma emission spectroscopy(GC-FPD/DCP).Appl. Organomet. Chem., 1989,3(4): 295-308.
[5] R. Compano, M. Granados, C. Leal, et al. Liquid Chromatographic determination of triphenyltin and tributyltin using fluorimetric detection. Anal. Chim. Acta., 1995, 314(3): 175-182.
[6] P. T. S. Wong, D. M. Whittle, Y. K. Chau,et al. Butyltin compounds in severn sound, lake huron, Canada. Appl. Organomet. Chem., 1994, 8 (4) : 385-391.
[7] A.D. Uhler, G. S. Durell, W. G. Steinhauer, et al. Tributyltin Levels in Bivalve Mollusks from the East and West Coasts of the United States:Results from the 1988-1990 National Status and Trends Mussel Watch Project. Environ. Toxicol. Chem., 1993, 12: 139-153.
[8] J. L. Gomez-Ariza,I. Giraldez, E. Morales, et al. Stability and storage problems in organotin speciation in environmental samples. Environ Monit, 1999, 1: 197-202.
[9] Q. F. Zhou, G. B. Jiang, J. Y. Liu. Small-Scale Survey on the Contamination Status of Butyltin Compounds in Seafoods Collected from Seven Chinese Cities. Agric Food Chem,2001,49: 4287-4291.
[10] S. Tsunoi, T. Matoba, H Shioji, et al. Analysis of organotin compounds by grignard derivatization and gas chromatography–ion trap tandem mass spectrometry. Chromatogr A, 2002, 962(122): 197-206.
[11] J. L. M. Vidal, A. B. Vega, F. J. Arrebola, et al. Trace determination of organotin compounds in water, sediment and mussel samples by low-pressure gas chromatography coupled to tandem mass spectrometry. Rapid Commun Mass Spectrom, 2003, 17(18): 2099-2106.
[12] J. Munoz, J. R. Baena, M Gallego, et al. Speciation of butyltin compounds in marine sediments by preconcentration on C60 and gas chromatography–mass spectrometry. Chromatogr A,2004,1023(2): 175-181.
[13] N. Cardellicchio, S. Giandomenico, A. Decataldo, et al. Speciation of butyltin compounds in marine sediments with headspace solid-phase
Micorextraction and gas chromatography-masss pectrometry. Fresenius J Anal Chem, 2001, 369: 510-515.
[14] R. B. Rajendran, H. Tao, T. Nakazato, et al. A quantitative extraction method for the determination of trace amounts of both butyl- and phenyltin compounds in sediments by gas chromatography-inductively coupled plasma mass spectrometry. Analyst, 2000, 125: 1757-1763.



































