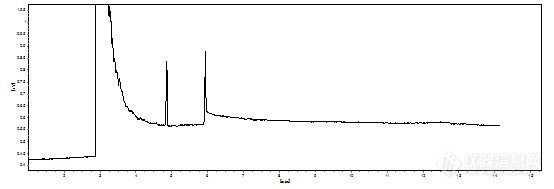
不好意思是我没说清楚,工程师维修的是FPD,我们在用FID,而且检测器和进样口的温度在美国药典上是270,200,我用的是280和250,不算低吧, 条件 初始 110℃ 30℃/min 至260℃ 保持10min 样品和内标全都在260℃出峰
至于气相条件在讲解的时候肯定是动了,比如载气流速什么的这些能显示出来的我全该回来了,分流那些因为之前处于摸方法的阶段,是不一样的,最有可能的就是隔膜了,尾吹应该没什么问题吧!
今天又要开始郁闷的一天了!!哎
qqqid(qqqid) 发表:以前是正常的。
“工程师来维修过检测器,顺便对气相大概讲解示范了一下”
维修完检测器有没有验证一下检测器的性能?应该让工程师现场做一下你的样品。
在工程师讲解示范的时候,有没有改变气相的条件?这个楼主需要好好想想查查。
另外,进样口温度是多少?是不是低了?