


省部重点实验室
第1楼2012/11/30
终于进入人体内部循环了,肝脏唯恐这些药物分子闯祸,用自己合成的白蛋白把这些药物分子装了起来。坐着观光车,药物分子在血液中就不会乱跑,而且在观光车上的药物分子也没有用武的余地。不过肝脏也不总是这么敬业,肝功能障碍时不舒服了他就会***,这样白蛋白的量就跟不上往常,同样量的药物,游离分子就增加了,闯祸的可能性也增加了。还有,会出现两种药物分子抢一辆车的情况。比如华法林,它是一种抗凝血药,通常他一个人搭车问题不大,但是遇到像磺胺类的药物分子,就跟他抢车,这样华法林就被挤下车,他的抗凝功能就会大大增强,肌体出血的风险也会加大很多。
随着血流,药物分子开始分布到全身组织。脂溶性强的分子分布的范围会大一些,反之水溶性的分子多半只能呆在血液里。药物分子所能达到的区域的大小,科学家用表观分布容积这个概念来描述。大脑是人体的司令部,因此里三层外三层裹得严严实实,称为血脑屏障,总之闲人免进,除了一些脂溶性特别好的分子。然而经过肝脏的一折腾,这样脂溶性好的分子也所剩无几。即便进去了,血脑屏障还有特殊的转运蛋白,叫P糖蛋白,它能把捣蛋分子遣返回血液循环,尽管花费不小要烧掉很多ATP。
药物分子这么曲折来到人体内部,很兴奋,开始找那些喜欢自己的蛋白,也是药物学家常说的靶点。这些蛋白都有自己的职责,有的在病原体身上,有的负责后勤转运,有的是酶参与化工生产,有的是细胞表面的受体,参与信号传递,等等。药物分子通常就在这些环节开始工作。其中最被药物学家津津乐道的叫G蛋白偶联受体,功能很多很强大。这些蛋白不喜欢药物分子赖着自己不走,产生共价反应,因为这是一件非常危险的事情,比如有机磷是一类毒药分子,他们喜欢与一个叫胆碱酯酶的蛋白抱在一起不分开。胆碱酯酶负责分解传递神经信号的分子——乙酰胆碱,没有了约束,乙酰胆碱就会持续兴奋神经系统,最后会导致呼吸麻痹,死掉!
有一个老牌的名药似乎可以例外,那就是阿司匹林。阿司匹林除了解热镇痛,还有活血化瘀、抗凝的作用,原因在于他可以抑制血小板的功能,血小板要想启动凝血程序,要靠环氧合酶运转提供血栓素等炮弹,但是阿司匹林可以和血小板里的环氧合酶共价结合,废除他的武功。本来这也是一件很危险的事,好在血小板的更新速度很快,用坏的血小板也会很快被我们清洁工巨噬细胞清除。
药物分子在体内循环着,一轮一轮的在肝脏转化,这些药物分子去了体内最后一站——肾脏。肾脏是个好员工,正常情况下只要水分充足,他会兢兢业业的把药物分子排到尿液里,除非管道堵塞结石,或者肾功能出了毛病。肾脏把药物分子排入尿液并非一视同仁。比如,如果同时服用了青霉素和丙磺舒,后者就会被优先排除,而青霉素就只能多等等了。
药物分子从体内排除,它的使命也就完成了。然而这段旅程,大家经历的时间可是有长有短,差别很大。有些几个小时就排干净了,有些可能要蓄积数年,比如一些农药。
省部重点实验室
第2楼2012/11/30
纸上谈药,一个畅销药的奋斗史(2)  [/url]
[/url]
——药状元的成才之路
本来我是打算写一篇关于药的科普帖子,可是写着写着发现时间不够,只好断开来个连载。好吧,既然一言难尽,就从今天开始,连载下去。我曾想过好几个写药的方式,不过顾虑很多,如果照药物的门类来写,我无异于在写一本药理书,实在是望尘莫及。既然这样我们就从故事开始吧。
每一个人都有自己的故事,每一个药也不例外;不过人越出名是非就多,药当然不例外。于是我也八卦一下从畅销药开始。作为一个畅销药压力自然不同于平常药。作为药物明星的它们,虽然卖的贵些,销量好些,给它的主人(药商)带来的收益大些,但是它无时不刻不担心着竞争者的攻击(山寨版仿制药,替代的新药),因为喜新厌旧对于人们对选择药物也通常不例外;另一个就是强大的舆论压力,万一出现什么不良反应新闻报道,那还不被口水淹死啊?
这是在现在。其实在很久很久以前,是没有什么畅销药的概念的。那时,人们都是凭经验到大自然中寻找药物,由于绝大部分是来源于植物所以我们也称为草药。起初,这些行为方式并不是为人类独有,其他的动物也知道吃“药”。人们主动的开发药物,得益于文化和文字的发展。从人群的经验中总结什么植物的什么部位经过怎样处理可以缓解怎样的症状,继而记载下来,慢慢的这就成了早期的药物数据库。但由于交通交流不便,那时主要的交通工具还是两条腿,因此每个地方的数据库版本也就不同。不过这并不妨碍它对治病救人的作用。
我一直以为,除了我们常教育孩子的“四大发明”,我们中国的中药其实也远远领先当时的欧美。那时的中国人做事很细致,热衷于建立各种数据库,一开始主要是经书史书,后来也包含了用药信息的医书。而且当时有文化的中国人普遍具有朴素的唯物主义精神,他们创造性的开发了“阴阳五行理论”,并把总结出的用药信息做了仔细归类。虽然华夏文明历经战乱,但是数据库多多少少还是保留下来了,尽管已经有些支离破碎了。
到明代中期,一个叫做李时珍的人出现了。原本他是一位大夫,在治病救人的过程中发现各个药物数据库都有些片面甚至错误。于是,他不辞劳苦,尽一己之力开始了一项伟大的数据库工程——修订《本草纲目》。这几乎耗尽了他毕生的精力。他去世的那一年,数据库刚好建完。随后这个伟大的数据库漂洋过海来到了日本。再后来,一个来中国访问的波兰人把这个数据库带到了欧洲。我认为这个数据应该算是源自中国的第五大发明。
当时,应该还没有美国,土著于美洲大陆的人还处于原始社会。而在欧洲,虽然科学的种子萌发得很快。人们终于赢了上帝一回,知道了并相信地球是圆的,而且是围绕太阳转的,大小两个铁球也会同时落地。不过对于人体与疾病的奥秘,他们知道并不比中国人多。很多时候,给病人放血成了一种很流行的治疗手段。
历史在静悄悄的流淌着,来自西方的科学家们用“科学”的思想,通过一个个精巧实验,慢慢发现了生命的奥秘,他们知道了心脏是怎样泵血的,而且知道这个机器会像柴油机一样燃烧葡萄糖,这个机器上还有很多像按钮一样的被称为受体的东西控制着机器的运转速度,而这些按钮的形状大小等等信息又被刻在了细胞核里的胶片上——DNA。而药物的开发,也在这些进展中悄然变化。这些变化渐渐开始决定一个化学分子是否能成为药的坎坷之路。
(2)现代药物的状元之路
我每次想到开发药物,就会联想到了古代科举考试的中状元。如今的畅销药其实也不例外!让我们追寻状元的经历看看吧——一个畅销药是怎样炼成的!
读过当年明月的《明朝的那些事儿》,多半知道中状元当首辅是多么不容易。好了,别吵了,开始我们的科举考试。
首先,必须说明朝还是颇具民主精神,加入这场角逐的考生资格并没有像以前的朝代有过多要求,不管你穷富帅丑,或者高矮胖瘦,只要有大明帝国的绿卡就好,另外不要有犯罪记录!
第一关是考秀才,以县为单位组织考试,难度不大主要是看基本功怎么样,初筛一遍脱颖而出的称为秀才;
第二关是乡试,秀才们从各自老家来到省城复考,考上的就被称为举人,原则上做举人就有当官的资格,而且享受免税的待遇;
再接再厉,跟着是会试,只有举人和那些“中央党校”(国子监)的毕业生才有资格参加,录取三百人。
最后一关是殿试,这是决定大好未来的最后一关。第一名就是我们说的状元,前程无忧。其他的称为进士,都还有机会。不过,最后能不能当宰相或者首辅,还有很长一段路,除了能力之外,运气也很重要。当上首辅也还不是终点,能否做个出色的首辅,纯粹的有利于人民的首辅,还必须满足两个方面。一是,要有才华,有卓越的行政管理能力;二是要有高尚的理想和良好的道德素质。不能把当首辅变成敛财的个人机会!
回到畅销药的成长之路,我们不难发现其中的坎坷和标准也有惊人的相似。
那么好吧,我们的畅销药开了漫长的征途!
首先,作为药物的候选化学分子,你们的机会是平等的,开始展示自己的才华吧。当然我是有要求的,这取决于你们的开发方向。比如,我要开发抗肿瘤药,我会先给你们一堆特定的肿瘤细胞,谁在单位时间内杀掉的最多,谁就可以胜出进入下一关。反之,只能OUT!出局!
当然,药物学家唯恐参与竞争的化学分子种类太少,他们除了把植物中的化合物翻了个遍,还与化学家一起发明了一种可以大量制造不同化学分子的方法——组合化学,简单说来就是把化学分子拆成几个部分,然后像拼积木一样把各个部分拼在一起,由于每个部分都有好几个选择,这样的搭配组合几乎是海量的!生物学家也不甘示弱,他们发现有些微生物会产生特定结构的多聚酮化合物,这些化合物是在一条布满合成酶的生成线上组装起来的,不同的酶负责不同步骤的组装,如果对其中的酶进行改造那么就可以改变酶催化反应的类型,应该就可以生产出很多不同结构的产物了。不过他们的策略不同于化学家,他们从这些酶的基因开始改造起,这个领域也被称为组合生物合成。
有了足够多样的化学分子,就意味着从中筛出“人才”的可能性就增大了。不过竞争也是更加的惨烈!另一方面,考生多了,考试的设置自然要高效合理,不然猴年马月才能干完呢?
实际上,考试的标准很早就制定了。那就是保罗埃尔利希(Paul Ehrlich)提出的——药物受体理论,他认为药物之所以起作用,其实是因为药物可以和某个东西特异性的结合在一起。而他所谓的某个东西就是后来说的“受体”。再后来,科学家慢慢了解——所谓的受体其实是那些可以和药物分子结合的蛋白,这些蛋白和相应的疾病有关。这时,药物的初筛就变成了射击游戏,谁能射到受体上并且不掉下来就能进入下一关!
这第一关杀伤力极大,能进入第二轮的实际上很少很少。
我们的第二关开始了。大家不要怕,我们这个关不搞淘汰,我们搞些培训,你们都很优秀很有运气的分子,将来都是有机会登上市场笑看江湖的,不过在这之前我要对你们做些改造,你们实际上可以做更加优秀!
于是,先导化合物的优化开始了。可是怎么优化呢?你怎么知道该把我们怎么改?
好吧,在了解怎么改之前,我们先接受点理论培训。先说“酶”,作为蛋白质的酶为什么只能催化特定得化合物参与化学生产呢?因为每个酶就像一把锁,只有特定形状的钥匙才能插进相应的锁孔,受体也是类似。可是怎样才能让钥匙更好用呢?对的——改!
你们做为筛选出来的优秀分子,身材自然是不需要大的改动了。那就在边边角角做些修饰吧,添上不同的化学基团。可是添在哪,添什么性质的基团,那是有讲究的。这个过程称为构效关系研究。
起初,由于受体长得啥样并不知道,药物学家都是凭经验改造这些候选分子,有时活性会更好,有时反而不行了,改来改去不知道什么时候是个头,一时间很是纠结。科学家总是有招!汉施-藤田方程(Hansch-fujita)登场了!
药物的生物学活性取决于药物分子与受体结合的紧密程度,而决定结合紧密程度的是化学分子的结构了。能够从初筛中脱颖而出,说明基本的骨架体型不能变了。汉施-藤田方程把化学分子结构信息拆成基本结构常数和其他参数。这里的其他参数包括化学分子是否有带电的基团,水溶性怎么样,有几个极性基团,这些基团分布的位置等等。用结构信息作为方程的右边,用药物的生物活性作为方程的左边,方程式写完了。
具体做法是这样的,药物化学家会根据骨架,事先合成出一堆类似的化合物,当然这些化合物变化的性质是知道的,然后用这些化合物测试它们的生物活性,得到的数据放在方程中,再根据各个化合物结构信息中包含的数值,算出影响结构信息的每个参数。这样药物学家就知道,那个位置安个什么样的取代基会对活性有帮助,那个位置的侧链过长会降低活性,等等。根据这些指导,他们会再做一批化学分子进行测试,反复下来,就可以得到一组活性相对较好的化学分子了。至此改造工作也就告一段落了。
当然现在的情况已经大不同了,一方面由于分子生物学的飞速进展,我们得以知道很多作为受体的蛋白质的分子结构,并且这些结构可以在计算机上以模型显示出来。即便不知道,我们也可以很方便的通过相似性比对(BLAST)在数据库中找到类似的结构,然后用电脑模拟。有了计算机的帮助,我们很方便快捷的“画出”我们理想中的化学分子。这块现在叫“计算机辅助药物设计”。
经过这段的培训,筛出的化学分子和经过改造的他们的兄弟姐妹允许进入了下一关。跟着的这一关分为两个环节,一个体内活性测试,一个是毒性测试。
实践是检验真理的唯一方法!药物好不好,疗效很关键。于是,人们利用小鼠大鼠等动物,复制出与人类相似的疾病模型测试药效。不幸的是,很多在体外有很好表现的化学分子在动物身上就是不给力,那就对不起了,只能OUT!毒性测试,就好比考状元时道德测试。有才无德的人往往是更危险的,药物也是如此。总的说来,药物的毒性测试过程非常繁琐,项目数之广时间之长令人叹为观止,当然投入的人力物力也是不计其数。无论怎样,这件事总归告诉我们——人品很重要,药品安全第一!
关于这一点,学药的人都会在很早的时候知道“反应停事件”,这是一个在药品安全环节把关不严的一个非常惨痛的案例。作为治疗妇女妊娠反应的药物,最终造成了三万个畸形儿的恶果。
如果作为化学分子,你顺利通过了这么严格的考验,那么恭喜你进入下一关!
这一关的考验也非常多,不过很多是比较程式化的,也不会轻易的被“OUT”,除非表现极差。这一关主要是研究候选药物分子的代谢情况,吸收,分布,转化,排泄,药物分子之间的相互作用,等等;同时,要给它们办“身份证”,怎么分析它的成分,怎么能从“人群中”鉴定出它;另外,给它们包装包装也是必须。还有一件事,就是设计工业化的生产流程,尽量做到省钱又环境友好!
顺利的跑完这些程序,就可以申请临床考试!不过能否成为真正的药物,仍需提醒一句,“上市仍未成功,考试还要努力!”
临床一期,二期,三期,四期实验开始了。这个过程非常漫长、苛刻和复杂,这同时也是药物开发企业最不愿面对的环节,因为从这一刻起,候选的药物分子每一天都要烧掉大把大把的钞票!而且OUT的可能性非常大,这意味着前面所有的努力都“Game Over !”
简单说来,这四期的考核,毒性的观察仍然是核心!考官说了:“药物要想通过临床测验上市,最重要的一件事就是安全,安全,还是***的安全!”一期试验里主要就是关注安全。选用正常人测试药物的耐受程度,也就是用相对大的剂量不能出现什么比较严重的毒副作用,剂量的依据来自于动物实验数据和人之间的换算比例;还有就是考察药物在体内的过程,这个就是我们第一期节目说的主要故事。
二期试验,主要是小范围内选取相应的适应症病人,测试药效,同时也要关注毒性,并且根据结果设计以后的用药剂量。这是因为药物剂量与药效之间并不是一条直线,药物剂量到一定程度后,药效也不再怎么增加了。同时,试验的时候还必须遵守“双盲对照”原则,不然很难客观的评价药物的真实效果,同时也避免“安慰剂”效应。因为,你如果忽悠病人这是特效药,即便是普通的糖片他也会觉得很有用,然后病情还真的有好转!我们《西游记》里的孙悟空就曾经这么忽悠过朱紫国的国王,而且还真的治好了他的“双鸟离分之症”。
三期试验,就是用前面摸索出的剂量在大范围的病人中测试,同样是双盲对照。
三期顺利过关,药物就可以正式上市了!上市后观察期,也被称为四期。在这段时间里,药物如果出现了很严重的不良反应事件,依然存在OUT的风险!
进入市场的药物要想成为畅销药,天时地利人和是少不了的!比如,禽流感那段时间,抗流感病毒药就卖的很火爆,这是天时;各个国家需要也不一样,比如降脂减肥药在欧美很畅销,到非洲有些国家就不同了,这是地利;人和,比如药物经销商的销售宣传策略。
即便是这样,也难免“谋事在人,成事在天!”畅销药的命运也是如此!也许是生命的奥秘实在太过高深,我们还没有完全读懂吧!
参考资料:
1.《药物分子设计》郭宗儒主编;
2.《计算机辅助药物设计》叶徳泳主编;
3.《药物评价学》刘昌孝主编
省部重点实验室
第3楼2012/11/30
纸上谈药,一个畅销药的奋斗史(3)
——No drug is good drug !
Robert F. Furchgott
引子:记得小时看武侠片,一般绝顶高手都会解码武功的最高境界——“无招胜有招”。其实最好的用药方式就是不用药。不过我们今天要谈到的“NO Drug”并不是不用药,而是一个传奇的分子——一氧化氮。 的确有不少畅销药就跟它有关。今年一月份,美国匹兹堡大学的陈丰原教授来实验室访问,有幸一起陪同吃饭。他是一氧化氮和心血管方面的专家,席间跟我们讲起了NO发现那段惊心动魄的故事,当天晚上我很想把这段故事记录下来,无奈水平有限,他说的一些外国人的名字我都拼不来没法动笔,只好重新慢慢收集资料做些功课,中间经过一个春节杂事很多也一直没下决心写。今天想起他讲的故事,开始第三次“纸上谈药”的历程吧。因为是故事,所以会有些“艺术加工”,虽然不是“纯属虚构”,但是“如有雷同,纯属巧合,敬请见谅”吧!
在正式开始故事之前,有必要提一下诺贝尔,这段且称为“NO前传”吧。
1898年12月,Alfred Bernhard Nobel 去世前不到两个月的一天,他给他的一位同事留言:“医生给我开的药竟然是硝酸甘油,难道这不是对我一生巨大的讽刺吗?”言外之意,医生居然给我吃炸药?!没错,Nobel发明了炸药,硝酸甘油就是其中的主要成分。不过在他之前还有其他的贡献者。1867年,Brunton发现亚硝酸戊脂的物质,可以缓解心绞痛,同时可以降低血压;1846年,意大利化学家Sobrebro发明了硝酸甘油,但这是一种极不稳定的能够引起爆炸的液体;1867年,诺贝尔发明了安全使用硝酸甘油的方法,他将硝酸甘油同二氧化硅混合在一起,这样液态的硝酸甘油就变成了半固体状硝酸甘油炸药;1879年,英国伦敦Westminister医院的William Murrell提出将硝酸甘油稀释后就可以转变成一种无爆炸性的物质,该物质可以作为心绞痛的药物。硝酸甘油随即成为一线药物,畅销世界,直到今天还是。尽管当初人们并不清楚其中的机理,但是丝毫不妨碍它的使用。就在诺贝尔去世的100年之后,即1998年,诺贝尔奖授予了揭开这个谜团的工作。冥冥中,经过一个轮回!
我们故事的主人公就是获得1998年诺贝尔奖的三位科学家。现在有请男一号,Robert Francis Furchgott 。Furchgott 生于1916年,1937年在在北卡大学修完化学,然后于1940年在西北大学获得生物化学博士学位。有时真是感慨没生在那个年代,拿个博士学位好像不用花太长时间,不像现在至少都是5、6年。在华盛顿大学呆了几年之后,逐渐有了点名气,随后去了纽约州立大学downstate分校医学中心做了药理教授。这也是他一生中工作时间最长的一个地方。直到2009年去世。他的工作单位在一个黑人聚居区,很偏僻,治安和环境都不是太好,在今天看来这绝对不是一个好的选择。相比今天大学生倾向去的“北上广”,那里大概相当于我们的二三线小城市,而且是经济不算很发达的。那个时候,各种内源性的化学物质相继发现,乙酰胆碱、肾上腺素、组胺等等。Furchgott 把主要精力放在了,研究这些物质对血管平滑肌的作用,也就是今天的体外药理实验。实验装置也很简单,把新西兰大白兔的主动脉取出来,剪成螺旋状的血管条,一头固定,另一头连着一台张力换能器,血管条泡在恒温的水浴槽中,然后往水浴槽里添加各种药物,观察血管的收缩反应。Furchgott每天就干这个事,乐此不疲,而且一干就是几十年,逐渐成了这个圈内的“权威”。
早在1953年,那时Furchgott还没有去后来的单位,他发表了首篇乙酰胆碱和组胺致兔离体血管条收缩的论文,然而整体动物静脉注射乙酰胆碱或者组胺却引发了血管舒张的效应。Furchgott坚持自己的实验重复性很好,而且观察无误。1955年,Furchgott在一篇综述中提出假说——血管平滑肌上也有两种胆碱能受体,跟肾上腺素的alpha和beta受体一样。尽管后来,有别的实验室发现了乙酰胆碱对离体血管的舒张作用,不过碍于Furchgott的权威,再加上乙酰胆碱对非血管平滑肌的作用是收缩效应,几乎所有的人都没有深究。
解铃还须系铃人。直到1978年一次偶然事故,谜团终于被Furchgott自己撕开。那天是个值得纪念的日子,5月5日。早晨一到实验室,Furchgott照例给助手David布置了实验任务,然后匆匆忙忙出了门。David那天可能是跟女朋友闹分手,女朋友一个劲劝他离开这个鬼地方,David心情不是很好,做实验时也心不在焉,不过教授布置的任务还得继续。下午,教授回来了,他给教授汇报结果。这次结果很奇怪,不应该这样啊?于是,Furchgott领着他重新核查实验细节,David显得有些慌张。很快,教授发现了问题所在,不过并没有责怪他,只是叫他明天按照今天的步骤重复一遍,严格的重复一遍,他点点头。
具体说来,Furchgott给助手David布置的任务是这样的。如图1。把血管条挂上实验装置后,先加入apha受体阻断剂,这样血管平滑肌上的alpha受体就被阻断剂封闭了。然后加入乙酰胆碱使得血管有一个预收缩的过程。跟着加入肾上腺素,血管平滑肌就开始舒张,这是因为肾上腺素对血管平滑肌上的alpha和beta受体都能激动,如果事先封闭了alpha受体,那么肾上腺素只能激动beta受体,表现出的就是舒张效应。也就是说血管平滑肌上beta受体介导舒张。
Furchgott在核查实验细节时,发现了David的两个失误。一个是David处理血管条标本时,并没有把主动脉剪成以往的螺旋状,而是直接挂在装置上;二是,David加药时弄错了,他忘了加入alpha受体阻断药,而且把肾上腺素和乙酰胆碱的加入顺序搞反了,更糟糕的是加乙酰胆碱时,肾上腺素并没有洗掉。这样出现的结果就是,肾上腺素使得血管收缩,而后加入乙酰胆碱,血管开始舒张!奇怪了!
图1
省部重点实验室
第4楼2012/11/30
一连三天,David做出的结果都是一样的。Furchgott很纠结,会不会是两种获取标本方式对血管有什么不同的影响?他让助手拿不同的两份标本去做组织学检测。经过一段时间努力,结果出来了,血管平滑肌的衬里——血管内皮有区别。螺旋式的血管条标本几乎见不到完整的内皮细胞,而血管环状的标本内皮细胞没有损伤。难道跟内皮细胞有关?Furchgott发现导致螺旋状血管条标本内皮损伤的原因可能跟操作过程中反复的摩擦有关。
经过大脑飞速的运转,一个绝妙的试验计划在Furchgott的脑中形成。很快实验就做完了,结果很漂亮。
具体说来,这个实验是这样的:
一是实验设想,内皮细胞有无是舒张效应的关键吗?为什么刺激内皮细胞可以引起血管平滑肌舒张?会不会是内皮细胞受刺激后释放某种物质扩散到平滑肌并导致其舒张?
二是实验设计,分为四步。第一,正常的血管环标本摩擦前后的比较;第二,正常血管环用胶原酶处理前后的比较,看化学损伤;第三,如图2,三个血管环标本,1号去除内皮,2号含内皮,3号是把前两个并排挂在一起,且内膜相对。这也是后来被广泛称道的“三明治血管灌流模型”。结果1号,没有舒张,因为没有内皮;2号,有舒张,因为内皮完整;3号也有舒张,说明内皮完整的那个血管条受刺激后释放了某个东西使得临近的血管也舒张。
第四步实验,Furchgott还观察了缺氧和有氧情况下血管条的反应,发现缺氧时,即便内皮完整,血管条也不舒张。多年之后,人们终于证明内皮细胞释放的就是NO,而NO生成需要氧气时,不得不感慨Furchgott当年实验的精妙,竟然如此“鬼斧神工”!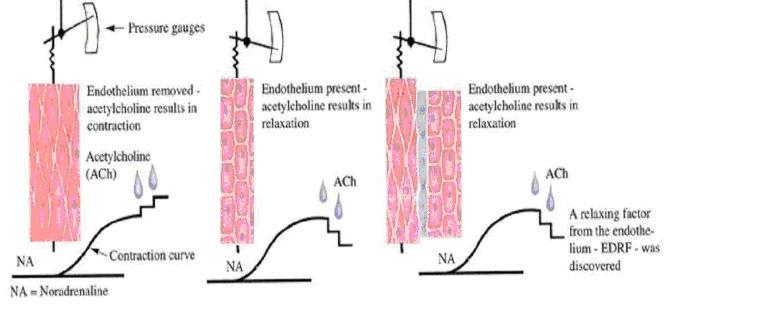
图2
拿到实验结论的Furchgott很激动,他决定把这个结果投到《Nature》。
题目是:“The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine”.内皮细胞是乙酰胆碱诱导动脉平滑肌舒张的必要因素。
Furchgott是一个很老实很本分的人,他一辈子也没玩什么先进的科研技术,要知道当时的分子生物学、转基因等等可是热火朝天啊。而他老人家几十年如一日的摆弄着“离体血管灌流装置”,虽然有些成就,可是激动人心的进展并不多。原本想着再干个几年就可以退休了。现在,他感到这些结果里面蕴含着惊天动地的秘密。所以,他终于决定试一试这个他以前都不曾想过的杂志。那时,投稿基本还是邮寄,Email还没有来得及普及。几周过去了,Furchgott 盼来了一封退稿信,两个审稿人的意见都是不予刊出,因为他提出的内皮细胞释放了某东西并不知道是什么玩意儿,太玄乎了。如果按照以前,退了就退了,老实的他不会多做争辩,换个杂志就是。可这一次,他再也忍不住了。他给编辑部写了一封信,请求编辑再另选一个审稿人。这一次他遇到知音,第三个审稿人正好也是做血管的,他看到Furchgott的结果很兴奋,随即通知编辑部接受了这篇稿子。
这时,那个内皮释放的东西还没有名字,正式有名字了是来源于一篇1982年PNAS的文章。在这篇工作里,Furchgott发现一种叫缓激肽的物质跟乙酰胆碱的作用类似,内皮释放的导致平滑肌舒张的那个玩意还是照例来了。Furchgott给这个蒙面者取了个名字叫“endothelium-derived relaxing factor”,即EDRF。
随后,EDRF的研究、血管内皮的研究开始风靡全球。EDRF的真实身份是什么呢?Furchgott很纠结也很兴奋,他孜孜不倦的工作着,虽然进展不多始终未能窥得EDRF的庐山真面目,不过根据蛛丝马迹,他猜测这是小分子的脂溶性很好的物质,根据硝酸甘油类的药物提示,有可能是NO。答案很快就会知道!
1986年夏季,美国实验生物学会在明尼苏达州的Rochester举行了一次研讨会,第一个报告人是Furchgott,他汇报了自己的工作进展,最后他说EDRF有可能是NO。第二个上台的是Louis J. Ignarro,他赞同Furchgott的观点,也认为EDRF是NO,并且拿出了一些侧面证据,比如含硝基的药物和乙酰胆碱诱导的EDRF都能使血管平滑肌中的一种叫cGMP的物质升高而不是cAMP,并且都出现血管舒张反应。此前cAMP作为肾上腺素的第二信使被发现的工作,让另一位美国科学家Earl Wilbur Sutherland 获得了1971年的诺贝尔奖,不过此时他已经辞世。
EDRF和NO的化学性质十分相似,例如EDRF性质不稳定,半衰期约是3-5秒,可以被超氧阴离子灭活,超氧化物歧化酶能使EDRF的半衰期延长到30秒。和血红蛋白(Hb)相互作用前,Hb的最大吸收峰在433nm, EDRF和血红蛋白(Hb)相互作用后, Hb的最大吸收峰移动到406nm。NO和Hb反应时也同样具有上述变化。Ignarro成了故事的男二号。
说者无意听者有心!台下有一位观众叫Salvador Moncada,来自英国。他的研究领域是阿司匹林和前列腺素等方面,此前他也曾猜测EDRF会不会是一种新型的前列腺素。现在他知道怎么做了,会议还没结束他就匆匆忙忙回到了英国,他要精确的证明EDRF就是NO,而不只是猜测!很快,他的团队做到了。他的精妙实验结果刊在了次年的Nature上。
具体说来,Moncada的实验是这样设计的。他把培养的血管内皮细胞吸附在微载体上,装柱后用缓冲液洗脱;洗脱液以不同时间间隔作用去除了内皮细胞的主动脉血管条以检测EDRF的生物活性(看血管条是否舒张),同时用化学发光法检测流出液NO的含量。并且把流出液与硝酸甘油的药理作用定量对比。结果发现,由缓激肽(BK)诱导内皮细胞释放的EDRF,不仅与NO在生物活性、半衰期等生物学特征上完全一致,而且可以被同样的药物阻断剂或激动剂抑制或增强!
非常完美的实验!随后,Ignarro也在PNAS上证明了EDRF就是NO,并且它还可以使cGMP升高。看起来,Ignarro好像晚了一步。
其实这时不应该忘记我们的男三号,Ferid Murad 。他是一位临床药理学家,大部分时间研究硝酸甘油的药理作用。他发现叠氮类化合物和硝酸甘油类药物能使血管平滑肌的cGMP大量生成,直接NO气体灌入作用效果类似,而cGMP是由鸟甘酸环化酶(GC)催化产生,如果事前抑制住GC,NO即使升高了,cGMP也不会增多。所以,整个故事就明白了。如图3所示。
图3
乙酰胆碱激活了内皮细胞表面的受体,通过一系列的信号传递,激活了产生NO的酶,NO生成后扩散穿越到下层的血管平滑肌细胞,激活了鸟甘酸环化酶,使得cGMP大量增加,最后通过信号传递产生舒张血管的效应。
随后,产生NO的酶也被发现了,主要贡献者是David Bredt 。不同领域的科学家还发现NO有别的功能。Michael Marletta 发现巨噬细胞释放NO,帮助巨噬细胞消灭病原体。John Hibbs发现巨噬细胞释放NO的前体物质是L-精氨酸。John Garthwaite 发现谷氨酸能刺激神经细胞释放NO ,似乎NO还参与神经信号传递 。David Bredt 再接再厉发现大脑里有一种特殊型号的NO合酶,即nNOS 。巨噬细胞里的NO合酶比较特殊,正常情况下很少,需要“战斗”时会增多,就是iNOS,发现者是陈丰原教授的同事,匹兹堡大学的Timothy R. Billiar 教授。
总之,NO越来越热。1992年,NO被Science评选为“年度分子”,并以“No News is Good News”为专题发表了专论,高度评价了NO的发现和伟大意义。终于,在诺贝尔去世100年后的1998年,诺贝尔奖殊荣给了“NO”。获奖者就是Furchgott、Ignarro和Murad 。消息一公布,英国人觉得很不公平,因为他们的英雄Moncada才是真正第一个证明EDRF身份的人,为此还写信给诺贝尔奖评委会,不过评委会给出的意见是:诺奖只能授予三个人,如果可以授给四个人,我们会考虑Moncada 。不过怎么说,上帝总归是公平的。试想想,如果Moncada不去参加那次会议,他还会改变自己的研究方向吗?从竞赛上说,的确是Moncada率先完成了任务。可是科学就是如此,提出想法的人总会得到优先尊重!
当然事情到这里还没有完。药物开发商辉瑞公司看到了其中的机会。硝酸甘油虽然继续畅销。但是它的药效时间很短,作为心绞痛的急救药还行,但是很难作为抗高血压的药物。辉瑞想从NO的道路里挖点“降压药”卖。很快他们把目标锁定在了cGMP身上。cGMP的半衰期很短,因为一个叫磷酸酯酶的蛋白会把它很快降解掉,这也是为了维持机体的平衡,持续的血管舒展会导致血压过低引发休克,人就挂了。当然如果是高血压病人,那就刚好合适。辉瑞针对磷酸酯酶开始漫长了开发流程。苦心人天不负,有一个化合物终于熬到了临床试验。不过,坏消息接踵而至。这个化合物的药效很差,在高血压病人身上几乎看不到什么降压效果,尽管还比较安全。
正在辉瑞绝望的时候,眼看数十亿美金的投入即将毁于一旦。然而柳暗花明又一村,好消息来了。一组身患糖尿病的40、50岁的男性病人很喜欢吃这个化合物,还主动问试验医生索要,尽管他们的高血压病没有什么改善。细致的调查员发现了其中的原因。这群病人常年阳痿,吃了这个化合物可以让他们“重振雄风”。食色性嘛,人之常情,难怪这群人很喜欢这个化合物。
辉瑞觉得这是个起死回生的机会,立刻改变药物的研发方向,把这个化合物做成“壮阳药”。这个决定也保证了辉瑞直至今天的辉煌。不多久,一个别名叫“伟哥(Viagra)”药物开始畅销全球。有了全球男人的“欲望”作保障,“伟哥”简直成了辉瑞的印钞机,销售额股票双双看涨!
原来,男性阴茎海绵体的磷酸酯酶是另一种型号,不同于别处。伟哥是这个型号磷酸酯酶选择性的抑制剂,cGMP在伟哥的干扰下,在阴茎海绵体积聚发挥舒张血管的效应,于是海绵体开始充血膨胀——坚挺。由于伟哥跟其他位置的磷酸酯酶结合得并不紧,所以对全身血压反而没有什么影响。天意如此,情何以堪!
当然,NO的故事远不止这些。后来发现,硝酸甘油在体内能变成NO,是由一个叫乙醛脱氢酶的蛋白催化的,即ALDH2. 而这个ALDH2本来是催化酒精代谢,让酒精的中间毒性产物乙醛变成无毒的乙酸。ALDH2有个突变位点,导致酶活力很差,所以ALDH2正常的人能喝酒,适量的酒精对身体的伤害不大。但是对于有突变的人,酒精简直就是***,大量的乙醛会阴魂不散。这种情况在中国人中还特别普遍。ALDH2的突变对于硝酸甘油的药效也同样适用。这也是临床医生常苦恼的问题,为什么硝酸甘油有的人用起来给力,有些人则不行呢。几年前,复旦的金力教授和卢大儒教授领衔的课题组,在中国人群中做了硝酸甘油药效与ALDH2基因型的关系,结果发在了《临床调查》(JCI)上。
与NO一同起步的血管内皮领域也搞得红红火火,甚至衍生出《内皮生物学》的分支。其中,日本的科学家做出了很大的贡献。他们发现了内皮素等多个血管内皮相关的因子。在上世纪90年代初期,还发现了血管内皮的修复干细胞——内皮祖细胞(EPC),这也是我现在博士期间的研究内容之一。
血管内皮细胞还分泌生长因子调节血管的生成,在癌症中很关键,正好硕士期间做过一点这方面的工作。肿瘤与血管新生又有另外一些故事,涉及到新的畅销药,我们改天再说吧!
参考资料:
1.唐小卿 PPT,http://www.docin.com/p-43707961.html
2.1998年诺贝尔医学或生理学奖.ppt
3.Robert F. Furchgott__The obligatory role of endothelial cells in the relaxation .pdf
4.Furchgott__ PNAS_Role of endothelial cells in relaxation of isolated arteries by.pdf
5.JPET__Association Between Cylic GMP Accumulation and Acetylcholine-Elicited Rela.pdf
6.Moncada__Nitric oxide release accounts for the biological activity of endotheliu.pdf
7.Moncada__Pharmacological Reviews__Nitric Oxide_Physiology , Pathophysiology , an.pdf
8.PNAS-1987-Ignarro-9265-9.pdf
9.Natue__Cloned and expressed nitric oxide synthase structurally resembles cytochr.pdf
10.PNAS-1993-Geller-iNOS_Molecular cloning and expression of inducible nitric oxide.pdf
11.http://www.mirm.pitt.edu/people/bios/Billiar1.asp#
12.PNAS-1990-Bredt-Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme.pdf
13.http://en.wikipedia.org/wiki/Robert_F._Furchgott
14.JCI__ (ALDH2) Glu504Lys polymorphism contributes to the variation in efficacy of sublingual nitroglycerin.pdf
精彩待续。。。。。。
省部重点实验室
第5楼2012/11/30
纸上谈药,一个畅销药的奋斗史(4)
——潜伏的诱惑 !
09年的一部《潜伏》,让国产谍战剧风靡一时,直到今天还没有消散。这些潜伏的无名英雄为今天的新中国做出了卓越了贡献。实际上这样的谍战剧,对于药物对于人体,时刻都在发生。说到这里,你大概知道我今天要说的话题。对,就是“前药”,也叫“潜药”。
那么什么是前药呢?如果你看过我写的第一章,大概还记得药物的体内旅程,药物分子要面临的其中一关就是肝脏,因为要在这里被代谢转化。或者说“解毒”,被“解毒”的药物分子多半就没有了药理活性,这个效应也被称为“首过效应”。药物分子就像冲锋的士兵,虽然斗志高昂,可是能冲过肝脏的防线,多半也都损兵折将。怎么办?“潜伏”起来!
也就是“悄悄的进村,打枪的不要!”
怎么潜伏呢?一般是把有活性的药物分子连上一个化学基团,让它失去活性,经过肝脏的代谢酶作用,药物分子正好可以变脸“现行”!
潜伏的确是个好主意!最早提出这个想法的是Adrien Albert ,一位来自澳大利亚的药物化学家。早年毕业于悉尼大学,后来在英国拿的博士学位,1954年当选澳大利亚科学院院士。Albert早期合成了很多抗疟疾和抗黄热病的化合物,还合成了一种名为Tacrine的药物,是一种胆碱酯酶抑制剂,用于治疗老年痴呆症(AD)。这部分以后再详述。Adrien Albert曾经就读的悉尼大学,还以他的名字命名一间药物化学实验室。
Albert在1958年的Nature上率先提出了“前药”的概念,文章题目是“Chemical Aspects Of Selective Toxicity”!此时,他的想法还只在于,可以利用药物潜伏的原理减少药物的毒副作用。
紧接着,1959年,Harper写了一篇综述,提出了“药物潜伏化(Drug Latentiation)”这个词,意思是——为了获得一个在体内酶作用下释放活性母体的新化合物,对该母体所实施的化学修饰。这便是最早的“药物潜伏手册”。
事实上,在这之前,几个已经热卖的药物都是“潜伏好手”。比如著名的阿司匹林。起初把它潜伏伪装的目的仅仅是因为它不好的口感和恼人的胃肠道刺激,没想到阴差阳错反而成就了经典。另一个案例跟“受体之父”Ehrlich有关,它就是“Arsphenamine胂凡纳明”,最早用于治疗梅毒。后来Voegtlin证实,胂凡纳明在体内经过代谢释放出活性分子氧分胂Oxophenarsine 。
好了,潜伏大戏开演了!不过在揭幕以前,我们还是要问几个问题。第一,为什么要执行潜伏任务?
这个问题显得有些愚蠢,对于干***,当然是为了更快的更好的取得***胜利,尽力减少一线战友们的牺牲了!对于药物,当然是更好的药效,更低的毒副作用!
第二,什么样的“谍报员”才算做优秀?
或者说一个优秀的谍报员应该具备哪些素质呢。首先,我觉得是坚定的信仰!“潜伏”的时候面临许多诱惑和考验,信仰不坚定者只能OUT!其次,是伪装的智慧!伪装的越真实,越是能迫近有价值的机密,当然也是保护自己的策略。再次是执行任务的能力!毕竟辛苦的“潜伏”不是来“打酱油”的!
所以对应的“前药”也有些类似,母体分子要确确实实能够发挥疗效,伪装的化学基团不要激起机体的反应,另外可以方便的在恰当的时机和地点脱去伪装!
好吧,“潜伏”的任务开始。
药物分子潜伏最多的任务是顺利通过胃肠道吸收。能顺利的通过胃肠道吸收入血并不是一件容易的事,至少对于大多数带电的极性分子是如此的。如图1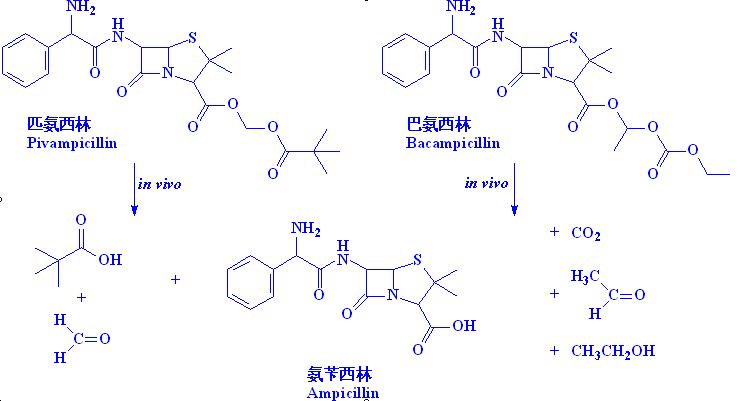
图1
氨苄西林是耐酸、广谱、半合成青霉素,虽然可以口服,但是吸收很差,同样的药物剂量只有注射给药时的20%-40% ,那是因为极性的侧链基团影响了药物分子穿越胃肠道的屏障。不过,修饰伪装一下就好了。可以把氨苄西林的侧链羧基设计成双脂前药,经过代谢酶的水解作用,释放出甲醛或乙醛以及氨苄西林,从而发挥药效。而对应的两个前药名字就是匹氨西林和巴氨西林。这二者在体外并没有抗菌活性。但是口服确实有效,说明预设的潜伏计划成功了!
这样的案例还很多!通过这种人为的修饰伪装,很多药物得以方便的吸收入血,充分利用!
药物潜伏还有一个重大任务就是“组织特异性的分布”。药物分子的表面上不可能安装GPS,没办法为它们引路,多数时候它们只能“随血逐流”。理论上说,可以流经体内每一个地方,除了大脑。
大脑的特殊之处在于它的屏障系统特别严密。绝大部分药物分子都只能望墙兴叹!而大脑需要的能量分子如葡萄糖氨基酸之类都是由特殊通道专门提供!
药物分子要通过怎样的“潜伏计划”才实现组织特异性分布呢?
任务很艰巨,成功者不多,不过也有例外。科学家把药物分子和一个叫“二氢吡啶”的载体连在一块,经过伪装的分子脂溶性很好,能够闯入血脑屏障进入大脑。图2.
图2
省部重点实验室
第6楼2012/11/30
图2
进入大脑后,前药分子被酶氧化带电从而变成水溶性的分子,所以只能进不能出,只得锁在脑内。而体循环中的前药分子被氧化变成水溶性以后,很快的从肾脏清除出去。这样看来,前药分子就好像具有了“脑组织的选择性”。
现在这样的“潜伏计划”已经得到了很广泛的实践。
另外一个选择性进入大脑的前药是——左旋多巴,它是神经递质多巴胺的生物前体,由左旋多巴和外周的多巴脱羧酶抑制剂相连,所以前药分子不能在外周脱去羧基,而只能在脑中脱去羧基,释放多巴胺,发挥药效。目前左旋多巴在治疗帕金森氏病(PD)领域是非常畅销的。
还有一个案例是来自另一个畅销药——洛伐他汀Lovastatin ,著名的降脂药。我们知道肝脏除了负责代谢药物之外,还是胆固醇和脂质代谢的调度中心。肝脏里一个叫“羟甲基戊二酰-辅酶A还原酶”(HGR)的蛋白是负责胆固醇合成的关键点。它的抑制剂自然就是很好的降脂药,不过不是洛伐他汀,而是它的活性代谢产物。也就是说洛伐他汀实际上是一个前药,刚好可以在肝脏中代谢成降脂的活性形式。恩,真是恰到好处啊!
药物的潜伏计划有时在于出其不意!这一点对于抗肿瘤药物尤其必要!起初人们认为肿瘤细胞就是恶性增殖的细胞,用抑制细胞增殖的药物应该可以抗肿瘤!于是,肿瘤化疗直到今天仍是一项比较常规的抗癌手段。不过,问题是,人体内除了肿瘤细胞,还有很多种类的人体细胞是可以且必须增殖的,如消化道的上皮细胞、造血细胞、毛囊细胞等等。那么怎么提高化疗药物对肿瘤的选择性呢?“潜伏”是一个选择。
起初的想法是,前药想办法让它毒性很小,经过肿瘤细胞特有的酶代谢激活,变成杀手杀掉肿瘤细胞。代表性的案例有环磷酰胺。
还有一个化疗药氟尿嘧啶也可以做类似伪装。氟尿嘧啶抑制肿瘤的机理是阻止肿瘤细胞DNA的合成。由于尿苷磷酸化酶在肿瘤组织的中含量明显高于正常组织,这就给潜伏提供了思路。脱氧氟尿苷(Doxifluridine)就是氟尿嘧啶的前药,与氟尿嘧啶相比,这个前药具有更高的治疗指数。值得一提的,骨髓细胞中没有这个代谢酶,因此潜伏者的骨髓毒性相对很弱。
不过对付肿瘤,武器的升级是必须的!潜伏一代不够给力了,潜伏2.0版本出炉。不过这主要归功于抗体导弹的诞生。具体做法这样的,有点类似《借枪》的情节。抗体分子先精确找到肿瘤细胞或组织,这一点是可能做到的。因为,肿瘤细胞再怎么隐藏,总有特异性的分子露在外面,如果找到这个特意性的目标,剩下的制备抗体导弹的工作也是可以进行的。
抗体导弹摧毁目标有两种方式,一是直接连上一个炸弹,比如放射性的元素,干掉肿瘤;
二是,连上一个酶,当然最好是人体内没有的酶类,比如产生细菌耐药性的“beta内酰胺酶”,这个酶是由耐药菌分泌的用于破坏抗生素分子结构的。细菌耐药本来是一件令人沮丧的事。但是这回却有意想不到的妙用。
杀伤肿瘤的药物分子事先与一个青霉素结构的分子相连。合并的“潜伏者”没有抗肿瘤活性,自然对正常组织也没有什么毒副作用。这时,先给机体输入抗体,抗体一头精确制导找到肿瘤细胞,另一头连接上的正是beta内酰胺酶。制导完毕后,沉默的杀手进场。尽管,前药分子可以分布到全身各处,但是只在有beta内酰胺酶的地方,也就是肿瘤所在的地方才被激活释放炸弹。总的过程可以描述为:寻找目标,制导定位,前药进入,定点爆破!默契的配合!见图3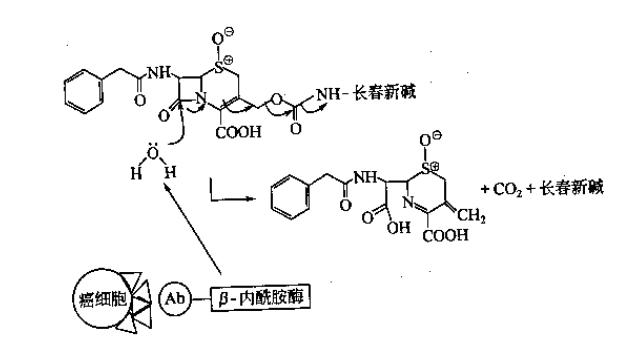
图3
潜伏的药物真好!可以解决很多问题。那么就没什么代价吗?
当然,有!
这里给的一个案例是抗血小板药物氯吡格雷,商品名波利维,制药巨头赛诺菲安万特的王牌畅销药。这个药常用于急性冠脉综合症ACS。
心脏一刻不停的给全身各组织泵血,而它自身消耗的血液是由冠状动脉提供的。由于饮食遗传年龄等等很多因素,冠状动脉会逐渐变得狭窄。管道拥塞,心脏自身的血液供应就成问题了,特别是在剧烈运动或者激动的时候,心脏加速血流却跟不上,这时就会心悸胸痛。
经皮冠脉介入治疗是一个比较直接的治疗手段,简单说来就是把狭窄的管道撑开,然后搭上一个支架。本来管道拥塞就常伴随着血小板的聚集,放入支架虽然扩充了管道,可是却会加剧血小板的聚集,引起新的堵塞。这是一个很麻烦的问题。
起初的方案是吃阿司匹林。没错,又是它!但是阿司匹林却很难控制,吃多了胃肠道受不了,吃少了又怕血小板重新堵上。
氯吡格雷就是直接针对血小板,准确的说是它的代谢激活物。因为,氯吡格雷是个实实在在的“潜伏者”。激活的氯吡格雷会与血小板细胞膜上的一个叫ADP的受体紧密结合,不分开(这种情况不多见)。被堵住的血小板也就被废了武功。好在血小板的更新还比较快!停药后,它们的正常功能还是可以恢复的。见图4.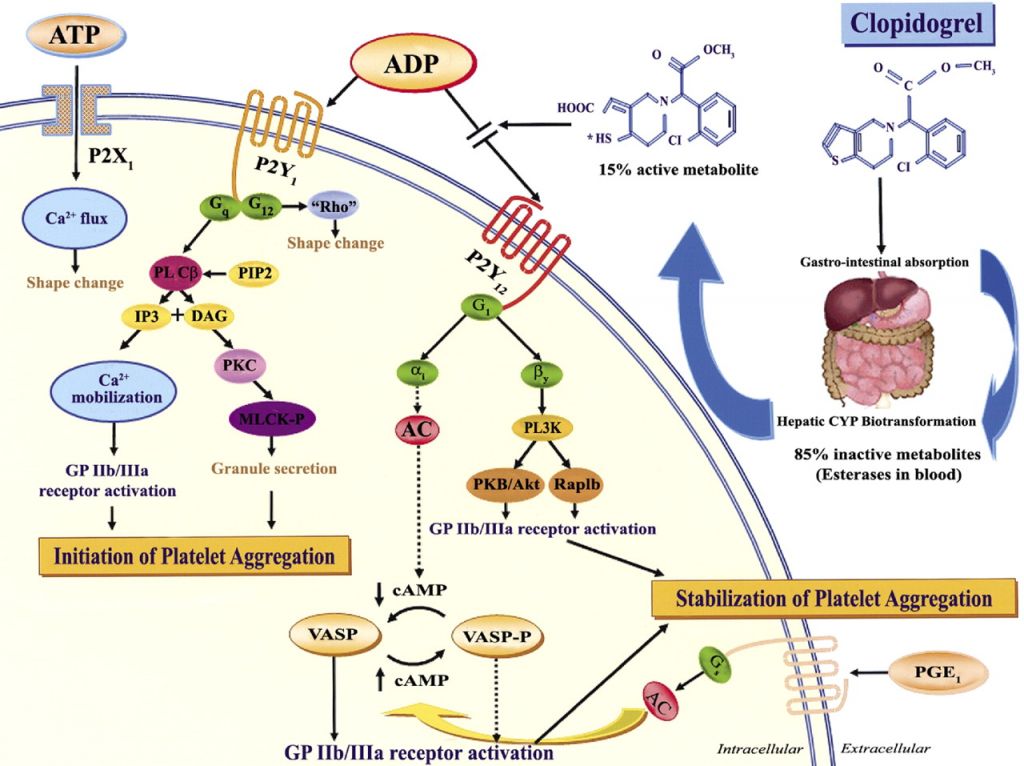
图4
氯吡格雷精妙的设计在临床试验和后来的市场上反应很好。由于现在人的不规律饮食,患ACS的病人和做PCI的病人非常多。氯吡格雷一时间成了赛诺菲的印钞机。
不多久,一些杂音陆续出现,有些病人即使使用了氯吡格雷仍然不能控制血管栓塞。这是为什么呢?很快,科学家发现,一个负责催化氯吡格雷激活的酶CYP2C19如果有突变,那么活性代谢物就不能顺利的产生,药效得不到发挥,这可能是问题症结所在。一些临床证据也陆续支持了这个结论。
这样,争议就来了!还要不要用氯吡格雷呢?有的说,不用吧,因为好些“非前药”的新型血小板抑制剂还在那等着呢。有的说,可以选择性的用,先给病人检测一下相关的基因型不就好了吗?毕竟,携带突变的病人并不是占了大多数,而且氯吡格雷经过这么多年的使用,安全性能还是了解得比较清楚的,况且检测的费用相对药费还是便宜得很多。
随着后基因组时代的到来,我们可以更加深入的了解我们体内与药物作用的蛋白,当然也可以设计出适合每个个体的独特的“前药”。
那一天还会很远吗?
参考资料:
1.李安良 主编.《生物利用度控制——药物化学原理、方法和应用》.化学工业出版社.
2.Nature__Chemical Aspects Of Selective Toxicity.pdf
3.JMPC__Drug Latentiation.pdf
4.王淑月PPT前药、硬药、软药.ppt
5.JAMA__Association of Cytochrome P450 2C19 Genotype With the Antiplatelet Effe.pdf
6.http://en.wikipedia.org/wiki/Adrien_Albert

































